有哪些高人做过1、糠醇 2、顺丁烯二酸酐 3、甲基丙烯酸甘油酯 4、苯磺酸 5、胺菊酯、氯菊酯 。它们的GC条件是什么啊?急等回音,谢谢!
请问从哪儿能查到海水中下面的杂质含量测定的方法:甲醇氯化石蜡-52乙二醇二乙二醇二甲基甲酰胺苯乙烯丙酮冰醋酸氯仿甲苯二甲苯1,4-丁二醇苯酚环氧乙烷
哪位同道有 对氨基二甲基苯胺比色法 GB 18056 –2000 附录A 用来检测甲硫醇的,急需!!!
有哪位大侠做过三(三甲基硅烷)硼酸酯或三(三甲基硅烷)磷酸酯的分析啊?用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的话选用何种柱子合适啊?
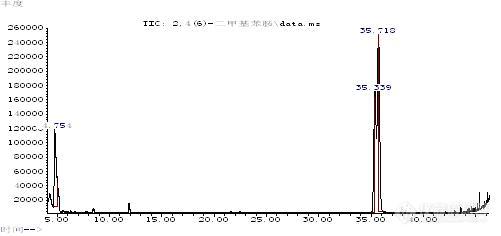
2,4-二甲基苯胺和2,6-二甲基苯胺的鉴别2,4-二甲基苯胺和2,6-二甲基苯胺同属于国家强制标准GB18401-2003附录C中所列的还原条件下染料中不允许分解出的23种芳香胺之一,二者又属于同分异构体,沸点和极性都很接近,故在检测过程中很难鉴别。目前,对于两者的分离鉴别主要靠液相色谱来实现,而使用气-质联用仪来鉴别两者还没有很好的方法。而针对有害芳香胺的气相色谱-质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。由于2,4-二甲基苯胺和2,6-二甲基苯胺沸点太接近,单纯依靠两者的沸点差异来实现其分离鉴别是有一定难度的。于是,作者考虑采用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),除了利用2,4-二甲基苯胺和2,6-二甲基苯胺的沸点差异外,再利用中等极性柱对于二者的保留作用差异来研究二者的分离鉴别。通过改善优化色谱条件,作者使用中等极性色谱柱DB-17MS,同时使用三阶程序升温,实现了2,4-二甲基苯胺和2,6-二甲基苯胺的较好分离。1 试验1.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)叔丁基甲醚 分析纯 国药集团化学试剂有限公司甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂2,4-二甲基苯胺和2,6-二甲基苯胺均为德国Dr.Ehrenstorfer公司。1.2 试样的制备分别称取适量的2,4-二甲基苯胺和2,6-二甲基苯胺,以甲醇为溶剂分别配制适宜浓度的2,4-二甲基苯胺溶液、2,6-二甲基苯胺溶液和2,4-二甲基苯胺和2,6-二甲基苯胺混合溶液。1.3 仪器操作条件色谱柱:DB-17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至[/font
氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
请教大家一下,一种用于固相萃取的试剂:二苯基硼酸二氨基乙酯(DPB或DPBEA)大家用过没有。我在文献上看到sigma公司有卖,可是我没有买到。有谁知道还有其他哪个公司可以买到,或者有其它的替代试剂没有。谢谢大家!!
有人有N,N-二甲基对苯二胺纯度测定方法或相关标准吗?谢谢了。
请教:相同浓度的异辛醇和邻苯二甲酸二异辛酯[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS分析,邻苯二甲酸二异辛酯峰正常,而异辛醇峰几乎没有?Finnigan MAT [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]Q Rtx-5MS柱子,温度60--280在上述条件下可以检测邻苯二甲酸酐吗?
大家好! 请问哪位大虾用过HPLC分离甘油,丙二醇,乙二醇,甲基丙二醇,TMP,新戊二醇,二甘醇等多元醇,具体方法能否告知,在此谢谢了!!!!
正丁醇和二甲基苯分不开,要求分好以后有四个峰,最好是填充柱.
请教各位大侠有没有做:臭氧危害物質(CFC/HCFC/Halon),氟溴烃,單甲基二氯二苯基甲烷,單甲基二溴二苯基甲烷 (DBBT),單甲基四氯二苯基甲烷 的检测方法,有的话能不能给我传一份,我的mail是wei.gao@isti.ocm.cn。万分感谢!
[color=#444444][color=#444444]求助,现在在做色谱检测这方面,要求分离苯甲酸,苯甲醛,苯甲醇,我用N-甲基吡咯烷酮作为内标物,溶剂为水。想问下N-甲基吡咯烷酮会与这三种物质发生反应吗?如果体系内有酸的情况下呢?[/color][/color]
对氨基二甲基苯胺光度法测定水样中硫化物的几个问题,希望那个大侠帮忙解决一下?我们公司使用的工艺是低温甲醇洗,测定的是其贫甲醇中的微量硫化氢含量?在测定过程中遇到的问题是:1、回收率达不到95%以上;2、用不同的稀释倍数测定同一个样品,其测定结果相差很大。3、贫甲醇中的硫化氢含量其实不多,但是把上述方法中药品加进去后,其颜色一直很深,且最后不会变成蓝色,但是用硝酸银定性,又感觉硫化氢很多。
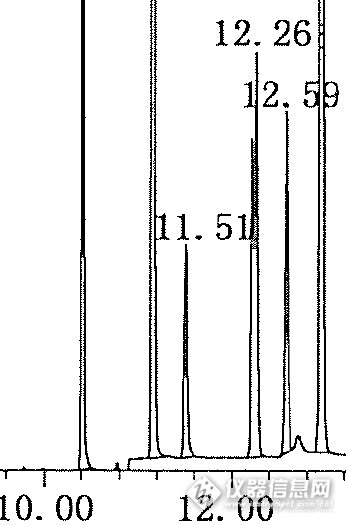
各位大侠好,昨天按照GB/T 21911-2008做了邻苯16P的方法,参数如下:http://ng1.17img.cn/bbsfiles/images/2015/02/201502270927_536552_2808019_3.jpg走完SCAN后,设置了SIM模式:http://ng1.17img.cn/bbsfiles/images/2015/02/201502270928_536553_2808019_3.jpg总离子流图如下:http://ng1.17img.cn/bbsfiles/images/2015/02/201502270930_536554_2808019_3.jpg发现第六个峰,也就是邻苯二甲酸二(4-甲基-2-戊基)酯有裂分(Ret time: 12.1):http://ng1.17img.cn/bbsfiles/images/2015/02/201502270933_536555_2808019_3.jpg然后我又看了一下标准,发现标准里的那个峰也是裂分的:http://ng1.17img.cn/bbsfiles/images/2015/02/201502270935_536556_2808019_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/02/201502270935_536557_2808019_3.png请问:1. 这是为什么?2. 我是积后面的那个峰呢还是两个峰积在一起呢?
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
有哪位大侠做过甲基硼酸的测试啊?谢谢指教!
0.1%甲基红乙醇溶液70ml0.1%溴甲酚绿溶液100ml1%硼酸溶液10L配置的吸收液,有悬浮物,应如何配制才能让该吸收液没有悬浮物?
1. 在用亚甲蓝测水中硫化物的时候,需要配置0.2%的对氨基二甲基苯胺溶液,我查了一下,他的别名也可以叫叫[font=宋体, Arial, Helvetica, sans-serif][size=12px]N,N-二甲基对苯二胺二盐酸盐,应该没买错吧。[/size][/font]2. 他的储存条件不是-20℃吗?这样它里面会不会含有很高的水分?那我称取2 g的 时候,需不需要干燥,还是直接就称取2 g 就好了。3. 还有就是配置好0.2%的对氨基二甲基苯胺溶液后,怎么储存呢?室温?冷藏?冷冻?如有回复,万分感谢!![img=,690,449]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239193272_1018_5383916_3.png!w690x449.jpg[/img][img=,690,299]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239240158_2694_5383916_3.png!w690x299.jpg[/img]
0.1%甲基红乙醇溶液70ml0.1%溴甲酚绿溶液100ml1%硼酸溶液10L配置的吸收液,有悬浮物,应如何处理?应如何配制才能让该吸收液没有悬浮物?顺序是怎样呢
有做水中硼酸的吗?现在买不到试剂(甲亚胺-H、甲基二戊醇等),有没有山东附近的试剂公司的联系方式。给一个,谢谢![em24] [em25] [em26]
配制完N,N二甲基对苯二胺,溶液应该呈紫色,为什么这次溶液呈透明淡黄色?
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
如题,发现这两个物质在DB-5的柱子上是分不开的,也看到有文献说液相可以分开的,试着做了一下,用甲醇和0.1%磷酸作为流动相也分不开,柱子是eclipse C18的,15mm长的。另:为什么标准中只限制了这两种二甲基苯胺,如果将其他异构体的二甲基苯胺误判为2,4-或2,6-怎么办?如何区分?
我想问一下怎么知道峰对应的是什么产物?甲醛,异丁醛,羟基新戊醛,1115酯,异丁醇,异辛醇,新戊二醇
[font=&][size=18px]N,N-二甲基乙酰胺又称乙酰基二甲胺、乙酰二甲胺,简称DMAC,是一种非质子高极性溶剂,有微氨气味,溶解力很强,可溶解的物质范围很广,能与水、芳香族化合物、酯、酮、醇、醚、苯和三氯甲烷等任意混溶,且能使化合物分子活化,因此广泛用作溶剂及催化剂。在溶剂方面作为高沸点、高闪点、热稳定性高、化学性稳定的溶剂,可用于聚丙烯腈的抽丝溶剂、合成树脂及天然树脂、甲酸乙烯酯、乙烯基吡啶等共聚物及芳烃羧酸的溶剂;在催化剂方面可用于尿素加热制氰尿酸、卤代烷与金属氰化物反应制腈、乙炔钠与卤代烷反应制烷基炔、有机卤化物与氰酸盐反应制异氰酸酯等过程。N,N-二甲基乙酰胺还可用作电解溶剂及摄影用成色剂的溶剂、脱油漆剂、有机合成原料、农药及医药原料。从C8馏分中分离苯乙烯的萃取蒸馏溶剂等。[/size][/font]
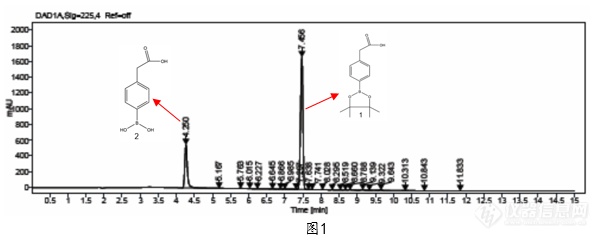
[align=left]近期我们遇到了一种硼酸酯类的化合物1,采用实验室通用方法进行检测的时候发现会出现一个很大的杂质2,根据工艺分析不可能会出现这么大的杂质,定量核磁检测发现该物质含量比较高,并不存在这个大的杂质,用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]去鉴定后发现该杂质为该化合物的水解杂质2(如图1)[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922409286_9676_5310417_3.png[/img][/align][align=center]图 1:流动相A: 0.05%TFA 流动相B: ACN条件下的样品色谱图[/align]为此我们判定肯定是检测方法出现了问题,首先我们排除稀释剂的影响,稀释剂为乙腈,做了相应的稳定性实验,发现临用新配情况下该杂质仍旧很大。由此我们判断可能是流动相导致该化合物1不稳定会水解生成杂质2。考虑到硼酸酯类化合物可能对酸不稳定,在酸性条件下会被催化水解成硼酸类化合物和相应的醇,因此打算更换其他流动相。首先我们尝试了碱性体系(如图2),由于该化合物1为酸性化合物,在碱性条件下保留较弱,但是从图谱可以看出水解杂质仍旧比较大,由此可以判断在碱性条件下该化合物1也并不稳定。[align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922406680_5272_5310417_3.png[/img][/align][align=center]图 2:流动相A: 0.1%NH4OH 流动相B: ACN条件下的样品色谱图[/align][align=left]随后我们又尝试了中性体系,采用中性体系的流动相进行测试(如图3)。从图3(a)可以看出,水做流动相条件下,由于流动相的离子强度不够导致峰形丑,还可以看出水解杂质2仍旧存在,但从(b)中可以看出当用乙酸铵作为流动相时候,峰形对称,水解杂质2也比较小。[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922412671_1242_5310417_3.png[/img][/align][align=left][/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922413707_3568_5310417_3.png[/img][/align][align=center]图 3:(a)流动相A: 水 流动相B: 乙腈 (b)流动相A: 10mM 乙酸铵水溶液 流动相B: 乙腈条件下的样品色谱图[/align][align=left]根据以上结果我们猜测:该化合物对酸碱都不稳定,但中性条件下只在乙酸铵体系下稳定,为此我们从化合物1本身及水解杂质2的结构分析,该化合物1中的硼原子为sp2杂化,还存在一个空的p轨道,这个空轨道易于接受水和醇等带有未共用电子对的亲核试剂的进攻而使硼酸酯水解([font='adobeheitistd-regular'][size=13px]其机理见方程式[/size][/font][font='dlf-32769-4-2073904376+zipdfa-8'][size=13px]([/size][/font][font='dlf-3-0-25052658+zipdfa-84']1[/font][font='dlf-32769-4-2073904376+zipdfa-8'][size=13px]))。[/size][/font]继续与水作用,生成相应的醇和硼酸。[/align][align=left][/align][img]" style="max-width: 100% max-height: 100% [/img]通过对此分析,似乎已经能够解释化合物1对碱不稳定的原因,即羟基中氧上的孤对电子会进攻硼的空轨道导致其水解,至于为什么在乙酸铵体系中是稳定的我们推测原因是乙酸铵的氮原子会与硼原子形成配对键,从而使该化合物1稳定。 虽然只是硼酸酯类化合物中的一种物质的检测,但是根据检测结果和分析可以为以后的该类化合物的方法开发提供思路,即通在对硼酸酯类的化合物进行方法开发时候,尽量不要采用酸碱体系的流动相,可以考虑用乙酸铵缓冲液作为流动相进行检测。[align=left] [/align]
我这个混合物中有邻苯二甲酸单辛酯,邻苯二甲酸酐和异辛醇。我用OV-17毛细管柱看不到邻苯二甲酸单辛酯,只有异辛醇,温度汽化298,检测(FID)260,程序升温180-260.请问是不是柱子没选对还是操作条件不对?那位高手指点一下,谢谢
DOP国标中,DOP的峰包括12.13.14这三个峰吗?(国标中注明:12、未知峰,13邻苯二甲酸二异辛酯,14、邻苯二甲酸二辛酯)辛醇有4种通素异构体,他们和邻苯二甲酸酯化都叫DOP吗?还是有其他名称?
最近做玩具增塑剂发现有一个样品出现了一个高浓度的尖峰,初步判断是邻苯二甲酸二正辛酯,但是与标物出峰保留时间不一致,标物质谱图离子是149和279,样品质谱图离子是149和279.1,丰度也在误差范围内,这样能说明样品就是目标物吗?因为标物是用丙酮色谱纯配制,而样品是用二氯甲烷分析纯提取的,这会影响目标物不同的出峰时间吗?我正在试用二氯甲烷色谱纯配标物,但发现二氯甲烷色谱纯进机会有很多杂峰,这要如何处理?







 我要推广仪器
我要推广仪器
 下载APP
下载APP