对甲苯磺酸盐在H谱上能显示峰么?
请教各位大侠:要如何检测原料药中的吡啶对甲苯磺酸盐残留量??
[em0912]把石油作为干扰组分(主要是芳香烃),测定石油中可能存在的重烷基苯磺酸盐(烷基苯磺酸盐的混合物,其特征吸收峰基本在272.0附近,所以作为一种物质处理),采用三波长法做工作曲线,目前大浓度重烷基苯磺酸盐没什么问题。由于石油中的重烷基苯磺酸盐含量可能很小,所以在小浓度时(石油200mg/L,磺酸盐1mg/L,正己烷为溶剂),数据没有重现性了!是不是到检测限了呢?想要增加石油的浓度,可是浓度大了吸光度曲线噪声太大,最大能做到200mg/L,有不能用普通的方法处理石油,怕应用中影响其中的重烷基苯磺酸盐含量?各位同行有什么建议,脑细胞都要用完了,嘿嘿!愁人啊?我的邮箱:Zhougd@jlu.edu.cn
有谁做过[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]双胍三辛烷苯磺酸盐??
萘磺酸盐多用于染料,是重要的中间体,萘磺酸盐化合物种类繁多,有一个磺酸基的,二个磺酸基的,三个磺酸基的。另外还有带羟基的、氨基的磺酸盐类。其含量分析多采用HPLC的方法,因为这类盐水溶性都很好,采用一般的反相色谱,其保留时间比较短,有文献报道用离子交换和离子对色谱分析居多。我分析过许多这类化合物,现做一些总结。 一般有三种分析条件,一是采用普通的C18柱或者亲水的柱,酸性流动相,几乎不含有机相;二是采用离子交换柱,如SAX;三是采用C18柱,但加入TBA之类的离子对试剂。从实际结果来看,采用离子对的方法最方便,分离效果最好。 采用第一种方法,是可以做的,我曾做过萘的1,2,3个磺酸根的盐的分离,在这种条件下,含3个磺酸出峰最快,几乎没保留,2个磺酸的有一定的保留,而1-磺酸保留时间比较长,但问题是3个磺酸的异构体根本无法分离,1个磺酸的化合物柱效很低,峰很宽,分析结果不太稳定。如果是带氨基和羟基的萘磺酸类化合物,用这样的方法就不行了,几乎分不开。当然不同位置的1,2,3个磺酸盐的分离是有差别的。实际分析中,很少有人用这样的方法做。采用第二种的方法,也是有文献报道的,我曾用NH4H2PO4为流动相,分离萘系磺酸盐,不同种类的无机盐分离的结果是有区别的,pH也有一定的影响,其分离主要靠盐的浓度来调节。其出峰次序是1,2,3个磺酸盐,同上面相反,不过在SAX柱上,峰形也不是很好,尤其3元酸,拖尾比较明显。采用第三种方法最常见,其离子对一般采用TBA居多,pH我一般调节在中性左右,流动相还必须加上一定的甲醇。如果调节好,峰形对称,保留时间恰当。不过在做的过程中,需要注意一些事,一是流动相平衡时间比较长,至少30分钟以上,二是同一方法条件,不同时间做,保留时间变化较大;三是,离子对的浓度要恰当,过浓会容易出现双峰的现象。在C18柱上,其出峰次序是1,2,3个磺酸盐。除了上面的方法外,还可以用戴安的PAX液相+离子交换柱来做,也有用毛细管电泳的方式。但在日常一般的分析中,采用离子对的方式是最普遍的。
组氨酸盐酸盐算不算管控的药品?
我想做组氨酸盐酸盐含量检测,没有什么好的方法,有哪位做这方面的请赐教。用液相,紫外分光光度等方法都可以。谢了! 邮箱:lei6657@126.com
请教甲基萘磺酸盐转化率的方法谢谢
各位大侠,我对[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]作的不多,可谓是菜鸟。我在实验中,需要将硫酸盐和氨基磺酸盐同时分析,但不知道怎样的缓冲液体系可以实现?希望大家指点。
近期部分地区质监局发文要求糕点食品生产厂家,送检原料“碳酸氢铵”中的“磺酸盐”。不知道为什么,检测标准是什么?
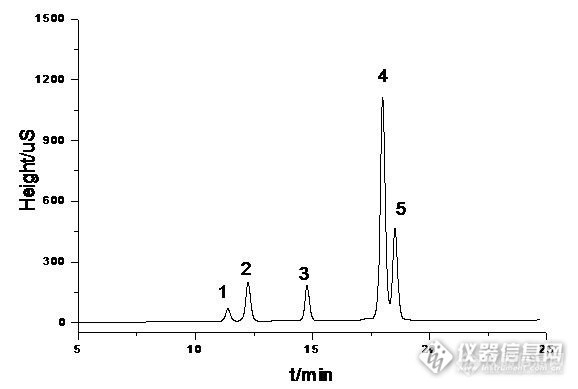
五种芳环磺酸盐的液相色谱分离方法浅谈 1-萘磺酸钠,2-萘磺酸钠,蒽醌1,5-二磺酸钠、蒽醌1,8-二磺酸钾和氨基萘酚-二磺酸钠五种物质均为强极性芳环磺酸盐化合物,在水溶液中完全电离成离子状态。在液相色谱分析中,五种物质的保留性质相似;因其为离子状态,C18反相柱对其没有保留,无法将其成功分离。本分析方法中采用离子对-反相色谱方法对其进行分离。具体分析条件和色谱分离图如下: 表1 色谱分析条件分析仪器U3000 HPLC 系统 色谱柱Diamonsil®(钻石)C18200*4.6mm,5um流动相A乙腈梯度洗脱:30minA: 20% ___ 45%B0.1%TBA+0.3%磷酸二氢钾pH6.5 温度30℃ 流速1mL/min 检测波长222nm http://ng1.17img.cn/bbsfiles/images/2016/09/201609141634_609777_3137073_3.jpg 图1 五种芳环磺酸盐的液相色谱分离图 1:蒽醌1,5-二磺酸钠,2:氨基萘酚-二磺酸钠,3:蒽醌1,8-二磺酸钾,4:1-萘磺酸钠,5:2-萘磺酸钠总结: 从色谱条件分析,本方法使用的是常规的反相C18色谱柱,在流动相中加入离子对试剂的方法增强保留性,用磷酸二氢钾调节pH至近中性。方法建立过程中发现pH和乙腈的比例对五种物质的分离影响较大,溶液的pH的大小影响芳环磺酸盐在水溶液中的分子形态,酸性溶液中,各物质形成成分子,中性和碱性条件下电离成离子状态,碱性溶液中有利于与离子对试剂的结合,但考虑到仪器和色谱柱不耐碱性能,选择中性范围较为合适。流动相中乙腈的比例对各物质在色谱柱中的保留时间影响。经过多次试验发现,梯度条件能较理想的将物种五种物质分开,而在等度条件下无法实现,特别是蒽醌1,5-二磺酸钠和氨基萘酚-二磺酸钠,1-萘磺酸那和2-萘磺酸钠两组性能保留性能相似的物质较难分开。 谱分离图来看,三种芳环二磺酸盐和萘磺酸盐完全分开,分离效果较好。蒽醌1,5-二磺酸钠和蒽醌1,8-二磺酸钾因空间结构差异,所表现出来的保留性有明显的区别,因此可以较好的分离。1-萘磺酸钠和2-萘磺酸钠结构性质非常相似,化学性质也相近,较难分离,目前还未建立将两者完全分离的方法。图中所示分离度不如其他物质理想。
伯胺的磺酸盐与伯胺在液相色谱上能分开么?
有没有人用液相来做全氟辛磺酸盐???现在都找不到具体的方法,还有配标样是怎么配的,要不要加酸?
汗渍色牢度用的L-组氨酸盐酸盐有几家供应商包装不一样,价格也有差别,大家一般怎么来技术验收的?
最近在做一个原料药,与磺酸成盐,手中的一份资料是醋酸缓冲液(PH5.70)和乙腈为流动相,但按此方法运行峰形拖尾,有个同事用0.1%三氟乙酸水和0.1%三氟乙酸乙腈作为流动相,峰形对称,但基线在220nm波长处有漂移和呈现波浪形的锯齿状态,查了下是因为三氟乙酸在220nm波长处有吸收,我接手后觉得应该把流动相PH值调整为碱性才对,因为药物成磺酸盐的话其本身应该是碱性物质,不知道我的想法还是同事的想法是正确的?这个原料在220nm和260nm都有吸收,合成人员非要用220mm,而三乙胺在220nm有吸收会导致基线很难平衡,请教高手碰到此问题如何处理?
一直听说磺酸盐类的对C18柱的伤害挺大的。想问下各位遇到测这类物质时如何维护C18柱的? 是不是做完后,用甲醇多冲冲?
如题,有机胺的结构与对甲苯磺酸反应成盐,结果发现对甲苯磺酸出峰位置与产物出峰位置一致,但是因为成盐的物质在乙腈里不溶析出,而对甲苯磺酸在乙腈中溶解性很好,所以可以判断是两个物质,但是现在想知道母液中产物到底占比是多少,怎么解决呢
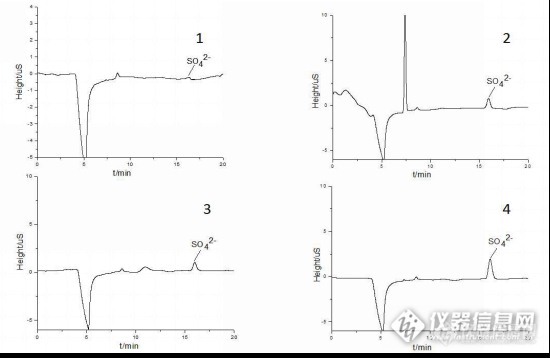
在线固相萃取-离子色谱法测定四种芳环磺酸盐中的硫酸根离子摘要:建立一种在线固相萃取离子色谱法测定四种芳环磺酸盐中硫酸根离子含量的新方法,将自装填的PGC-SPE柱应用于离子色谱系统对样品进行在线前处理,样品经过PGC-SPE柱处理后进入500μL定量环,通过阀切换-大体积进样模式使硫酸根进入阴离子检测系统。固相萃取流路以1.5mmol/L Na2CO3在0.8mL/min的流速对基体在线富集;分析柱采用SH-AC-3 (4.0×250mm) + SH-AG-3 (4.0×50mm),在6mmol/L Na2CO3 +4mmol/L NaHCO3条件下等度洗脱,柱温为35 ℃,流速为0.8mL/min,进样量20μL。结果表明:硫酸根离子在0.50-10.00 mg/L浓度范围内呈良好的线性关系,线性相关系数为0.9993,保留时间、峰高、峰面积的RSD均在0.28%~2.86%之间,方法检出限为0.0106mg/L,回收率在93.33%~105.59%之间;该方法具有良好的线性和重复性,整个在线分析过程在25min之内完成,进样量少,快速、高效。关键字:在线固相萃取,离子色谱,多孔石墨化碳,自装填技术,硫酸根离子1 前言多孔石墨化碳(PGC)作为一种新型色谱固定相,因其表面为纯碳结构、平整且无接枝的功能基团而具有耐强酸强碱、耐高温、性能稳定的特点,广泛应用于液相色谱和离子色谱中。PGC是二维结构、表面完全非极性且有可自由移动的π电子,因此与它化合物之间存在着疏水作用和电子间作用等多重作用力,从而在一定条件下对极性化合物、非极性化合物、异构体、聚合物等都具有保留性且对强极性化合物表现出强保留性。在样品前处理特别是复杂样品前处理过程中,通常利用固相萃取对样品进行净化与浓缩富集,除去干扰性的杂质,从而提高待测物分离度并保护色谱柱。Thermo公司首先推出以多孔石墨化碳填料为固定相的hypercarb 分析柱。在固相萃取技术应用中已有商品化的离线多孔石墨化碳固相萃取小柱(PGC-SPE柱),主要应用于液相色谱和离子色谱的样品的富集和基体消除。 萘磺酸盐和蒽醌磺酸盐属于多苯环磺酸盐,易溶于水的强极性离子型化合物。目前商品化的离子色谱固相萃取前处理柱,如:Onguard RP柱、H柱、Na柱以及聚合物树脂固相萃取柱等对非离子型有机化合物和常规离子基质有很好的前处理效果,但对芳环磺酸盐类的离子型强极性有机化合物保留性差,前处理效果不理想;在测定这类物质中残留的无机离子时,若不能很好地对该类物质进行基体消除,其进入分析系统后易保留在离子色谱柱上且较难洗脱,对分析柱造成损害。PGC因其特殊的表面疏水性质和带电性质使得它对该类强极性离子型化合物具有很强的保留性。本实验创新性的提出自装填可再生的多孔石墨化碳固相萃取柱并将其应用于在线离子色谱分析系统对四种芳环磺酸盐中的硫酸根离子进行测定,自装填和可再生重复使用的特点很大程度上降低了实验成本,在线基体消除较离线前处理在能保证理想的前处理效果和样品分析重复性的同时,能很大程度的减少样品污染并缩短了分析时间和样品量,更有利于复杂样品中阴离子的快速、准确的分离分析。2 实验部分1.1 仪器与试剂实验所用仪器为自组装离子色谱系统,主要部件包括:Ultimate3000 WPS-3000TSL自动进样器,ICS5000+紫外检测器,ICS5000+(SP/DP)双泵,ICS5000 TCC柱温箱(含一个六通阀和一个十通阀),ICS3000(SP)单泵,ED50A电化学检测器(DS3,带控温)Chromeleon6.8色谱工作站,(美国赛默飞世尔科技有限公司);WLK-6A阴离子抑制器(青岛仪趣仪器有限公司);AL-10电子天平(梅特勒-托利多有限公司);Milli-Q Advantage A10超纯水机(Millipore);BRANSON 2510超声清洗仪(BRANSON); 硫酸根离子标准储备液(100 mg/L,上海计量测试技术研究院);碳酸钠和碳酸氢钠(分析纯,99%,上海凌峰化学试剂有限公司);氢氧化钠 (w/w=50% ,默克公司);浓磷酸(分析纯,国药集团化学试剂有限公司);甲基磺酸(99.5%,上海阿拉丁有限公司);甲醇,乙腈(色谱纯,上海星可高纯溶剂有限公司)。 1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾(上海翰思化工有限公司提供)1.2 标准溶液的配制 分别取0.50,1.00,2.00,5.00,10.00 mL的SO42-标准储备液于100 mL容量瓶,超纯水定容后得到0.50,1.00,2.00,5.00,10.00 mg/L的SO42-标准液。 准确称取0.0242g、0.0213g、0.0248g、0.0257g的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾于50mL容量瓶,超纯水溶解定容,作为待测样品。1.3 色谱条件在线PGC-SPE柱为本实验室自装填前处理小柱(3.0×30mm),PGC填料粒径:30μm,阴离子分析柱: SH-AC-3 (4.0×250mm,9μm) + SH-AG-3 (4.0×50mm,12μm) (青岛盛翰色谱技术有限公司),柱温:35 ℃,流速:0.8mL/min;进样量20μL;定量环:500μL;淋洗液:6mmol/LNa2CO3 +4mmol/LNaHCO3; 分析时间25min;1.4 On-line SPE-IC系统的构建 On-line PGC-SPE-IC系统构建如图1 所示,实验建立了阀切换-大体积进样抑制电导离子色谱方法测定四种芳环磺酸盐中的硫酸根离子。系统中泵1用于在线固相萃取和进样,泵2用于阴离子分析,泵3用于SPE柱的在线清洗再生;利用一个十通阀和一个六通阀实现样品的在线固相萃取、大体积进样和在线前处理柱的再生,抑制电导检测器对硫酸根离子进行检测。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131609_609526_3137073_3.jpg 图1 在线SPE-IC系统2 结果与讨论2.1 PGC-SPE柱的装填和活化及柱容量 实验中所使用的SPE柱为本实验室自装填前处理小柱,采用湿法装填方法,以乙醇为分散剂,将填料以流动相的形式利用高压泵进行装填,装填过程先是填料在低流速低压条件下缓慢泵入柱体内,一段时间后再缓慢将流速调节为1mL/min装填1h以上,最后加大流速在高压条件下将填料压实;装填好的SPE柱压力约为120 psi。将装填好的PGC-SPE柱进行一下酸碱活化过程:酸冲洗(100mmol/LHCl, 1mL/min,30min)——水冲洗(1mL/min,20min)——碱冲洗(100mmol/LNaOH, 1mL/min,30min)) —— 水洗(1mL/min,20min)。酸碱冲洗的作用在于对小柱进行活化同时溶解填料中本身残留的一些酸碱可溶性杂质,避免其进入色谱系统损害色谱柱和抑制器。 利用紫外检测基线突越的方法测定PGC-SPE柱对该四种化合物的柱容量。分别配置500mL浓度为230.0、236.4、129.0、258.0mg/L的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾溶液作为流动相,以0.1mL/min的流速流过SPE柱,记录基线开始采集和发生突越的时间间隔,根据公式计算(柱容量=物质浓度*流速*时间)得:1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾的柱容量分别是0.8912mg,0.8307mg,0.2354mg和0.4373mg。2.2 色谱分析条件的选择2.2.1 硫酸根在SPE柱上洗脱条件(泵1淋洗液) 在中性条件下, 由于电荷的相互作用,PGC-SPE柱对硫酸根具一定的保留性,而酸性或者碱性条件下将其洗脱;实验中考虑到仪器系统兼容性和耐碱程度,选择低浓度碳酸钠作为洗脱液。由于使用500uL定量环大体积进样方式,流速为0.8mL/min,则SO42-的峰展宽必须控制在0.625min之内。根据实验结果可知(见图2),在0.5mM,1mM,1.5mM碳酸钠条件下,阴离子完全洗脱时间分别是1.10-2.20min,0.75-1.45min,0.65-1.20min;1.5mM浓度下的硫酸根离子在0.55min之内洗脱,因此Pump1选择1.5mM碳酸钠作为洗脱液。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131601_609519_3137073_3.jpg 图
[b][font=微软雅黑]【序号】:[/font][font=微软雅黑]1[/font][font=微软雅黑][/font][font=微软雅黑][font=微软雅黑]【作者】:辛若竹[/font][font=微软雅黑]1丁梅1李志远2石金娥3[/font][/font][font=微软雅黑][/font][font=微软雅黑][font=微软雅黑]【题名】:超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url][/font][font=微软雅黑]-串联质谱法测定水果蔬菜中双胍三辛烷基苯磺酸盐残留量[/font][/font][font=微软雅黑][/font][font=微软雅黑]【期刊】:分析试验室[/font][font=微软雅黑][/font][font=微软雅黑][font=微软雅黑]【年、卷、期、起止页码】:(录用定稿)网络首发时间:[/font][font=微软雅黑]2023-02-14 16:54:32[/font][/font][font=微软雅黑][/font][font=微软雅黑][font=微软雅黑]【全文链接】:[/font][font=微软雅黑]https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C45S0n9fL2suRadTyEVl2pW9UrhTDCdPD66OzI2bu69oZMPcJkM-v_Lpw6TNTOBPdNy0M811rYJFeVe7Mytt9cED&uniplatform=NZKPT[/font][/font][/b]

最近做氨基酸检测,用OPA-MPA衍生后,液相色谱荧光检测,c18的柱子其他氨基酸都分得挺好,就是组氨酸和甘氨酸完全分不开,就看到一篇中文文献里也是这个情况。我估摸着是哪个细小环节没处理好,有经验的大虾指点下缓冲盐试过磷酸盐和乙酸盐的,各自都加了四氢呋喃,乙腈,以及3乙胺等试过,走梯度,都是一样的,2个分不开。附图,大家看看,红圈的地方就是甘氨酸和组氨酸的出峰时间http://ng1.17img.cn/bbsfiles/images/2011/06/201106251547_301445_1642776_3.jpg谢谢
各位大哥大姐,请问大家有没有苯丙氨酸甲酯盐酸盐的手性分离条件?
现有个样品需要检测,但是用公司常规方法检测部出来,试了一些方法还是不行,用的流动相是乙腈和水;甲醇和水,pH值大约都在5-6,现请教下名称:H-Leu-OBzl.Tos-OH中文名称:L-亮氨酸苄酯对甲苯磺酸盐对于测这类产品,流动相的pH值有没有影响,能否提供下方法啊,急啊谢谢给位老师了啊
在检测甲磺酸酯类物质时,根据欧洲药典方法,选用碘化钠和硫代硫酸钠作为衍生试剂,其中对照溶液的配置方法是称取一定量甲磺酸酯类到5ml容量瓶中,加甲苯稀释定容摇匀,请问甲苯的作用是什么呢
有谁跑过盐酸组氨酸的薄板,对照溶液为自身样品溶液(50mg/ml)稀释而成(0.1mg/ml),以正丁醇-丙酮-浓氨溶液-水(10:10:5:2)为展开剂。显色后发现对照的斑点位置高于供试品。按说自身对照的话,Rf值应该是一样的啊,请问各位大虾,这是怎么回事啊?[em53] [em53]
现在手上有一组氨酸原料药,几乎相当于纯品。老板要我做组氨酸的有关物质,除了其他的氨基酸,其他杂质是什么无从下手。DAD,CAD检测几乎没有杂质,进样量40ul才有点杂质峰出来。手上有一台q-e,不知道能否先直接分析组氨酸原料药里的组分,然后再寻找合适的液相条件开发有关物质的分析方法。由于原料药杂质含量太低了,但是的确有,实在不知道怎么进行了,哎
原料药氨磺必利的起始物料阿米酸合成中用到对甲苯磺酸,在合成的过程中,可能会产生对甲苯磺酸酯,对甲苯磺酸酯为遗传基因毒性杂质,使用什么方法可以检测?有没有版友做过类似的检测,帮忙分享一下,谢谢!
本人在8月发表的一篇原创中提及”甘氨酸与组氨酸无法分离“的问题,在经过10多天的准备,已有不小的收获,现在分享。摘要 目的: 建立用高效液相色谱法测定人凝血因子VIII中氨基酸含量。方法: 采用6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 为衍生剂,与氨基酸柱前衍生后,用Agilent 1200 高效液相色谱仪,AccQ·Tag C18柱( waters 150 mm ×3. 9 mm,4 μm) ,以水Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液和乙腈进行梯度洗脱,检测波长为248 nm,柱温37 ℃,进样量10μL。结果: 各氨基酸在32 min 内测定完毕,回收率为98.7% ~ 101.5%。RSD 均小于1. 5%。结论: 本法分离度好,快速、简便,可作为产品的质量控制方法。关键词: 6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯; 人凝血因子VIII; 甘氨酸; 衍生物; 梯度洗脱; 高效液相色谱法;氨基酸; 含量测定人凝血因子VIII,本品对缺乏人凝血因子礓所致的凝血机能障碍具有纠正作用,主要用于防治甲型血友病和获得性凝血因子Ⅷ缺乏而致的出血症状及这类病人的手术出血治疗。该药物制备过程中使用了氨基酸( 精氨酸、丙氨酸、甘氨酸、组氨酸、盐酸赖氨酸、脯氨酸 等) 做稳定剂,为了保证药品质量和用药安全,应对其中氨基酸的含量进行控制。该法依据过量的6 - 氨基喹啉基- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 在一定条件和氨基酸形成稳定的衍生产物( 柱前衍生) ,用高效液相色谱法测定衍生产物,根据衍生产物的含量计算人凝血因子中各氨基酸的含量。1 仪器和试药1200 高效液相色谱系统( 美国Agilent 公司) ,配置低压四元梯度泵、1314B 紫外吸收检测器、自动进样器、柱温箱、Chemistations 化学工作站; Sartorius CP225D 电子微量天平( 德国Sartorius 公司) ; SartoriusPB - 21 型pH 计( 德国Sartorius 公司) ; LDZ5 -2 低速自动平衡离心机( 上海医用离心机厂) 等。各标准品均来自于中国食品药品检定研究院2 色谱条件及系统适用性试验色谱柱: Waters AccQ·Tag C18色谱柱( 3. 9 mm ×150 mm) ; 流动相: 水为溶剂D,Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液( A) - 乙腈( B) - 水( D) ,柱温:37 ℃; 检测波长: 248 nm。精密量取对照品溶液与供试品溶液10 μL,分别注入液相色谱仪,记录色谱图32 min。3 溶液制备3. 1 Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液称取三水乙酸钠190. 4 g,加注射用水1000 mL,搅拌,溶解,用稀磷酸将pH 调至5. 2,加入乙二胺四乙酸二钠溶液( 称取乙二胺四乙酸二钠100 mg,加注射用水100 mL,摇匀使其溶解) 10 mL,加入叠氮化钠0. 1 g 及三乙胺23. 7 mL( 17. 2 g) ,用稀磷酸滴定至pH 4. 95,用0. 45 μm 的滤膜过滤,于4 ℃储存,备用( 此条件下可保存6 个月) 。量取该溶液100 mL,加注射用水稀释至1000 mL,混匀,即得Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液。3. 2 对照品储备液混合对照品储备液精密称取各氨基酸对照品适量,置同一100 mL量瓶中,以注射用水溶解并定容至刻度。制成含氨基酸含量均含5. 0 mg·mL - 1 的混合对照品溶液,即得。单个对照品储备液: 精密称取各含氨基酸的各对照品适量,分别置100mL 量瓶中,用注射用水溶解并定容至刻度。制成分别含各氨基酸的单个对照品溶液,即得。3. 3 供试品储备液3. 3. 1 加样回收率试验溶液精密称取各氨基酸各0. 3200,0. 4000,0. 4800 g 和辅料适量,加人凝血因子VIII原液7. 5 mL,肝素钠适量,用1. 0 mol·L - 1 盐酸调pH 至6. 9,加0. 01 mol·L - 1枸橼酸三钠溶液溶解并定容于20 mL。分别制备成16. 0, 20. 0, 24. 0 mg·mL - 1溶液。3. 3. 2 空白溶液 按公司处方,加入辅料的混合物,用注射用水制备各空白溶液3. 4 内标溶液精密称取α - 氨基丁酸( AABA)0. 4 g,加注射用水定容至100 mL。4 氨基酸衍生方法4. 1 精密量取供试品储备液、样品及对照品储备液各1. 0 mL,加1. 5%磺基水杨酸9. 0 mL,混匀静置2 h以上, 3000 r·min - 1离心10 min,留取上清液。4. 2 精密量取“4. 1”项下上清液1. 0 mL( 其中对照品储备液对应上清液分别精密量取0. 06, 0. 4,0. 8,1. 0, 1. 2, 1. 6 mL) ,分别置10 mL 量瓶中,用注射用水定容。制备成供试品溶液、样品溶液及浓度分别为1. 5, 10. 0, 20. 0, 25. 0, 30. 0,40. 0 mg·mL - 1 的对照品溶液。4. 3 精密量取“4. 2”项下溶液各100 μL,分别加注射用水0. 4 mL 及内标溶液20 μL,混匀备用。4. 4 精密量取“4. 3”项下溶液30 μL 放入衍生管中,加硼酸缓冲液( pH 8 ~ 10) 210 μL 涡旋混合,并加入AQC 衍生剂60 μL 涡旋混合15 s,即为各供试品溶液,待用。
求分离对甲苯磺酸,邻甲苯磺酸,间甲苯磺酸的方法。用的是安捷轮1200,c18柱。谢谢了[em0812]
首先说一下,我不是搞分析的,有些基础概念可能不清楚,请大家见谅!最近实验室想分析一个水样,推测里面主要含有苯磺酸钠、羧酸钠、及无机盐(NaCL),问别人说可以先做一下液相色谱,看看是不是能分得开,下来再作[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url],分析一下!但是,我咨询一些分析测试中心,我一点也不懂,还以为给人家就可以做了!他们都要求我提供分离条件??我晕所以在这,请教下各位,那位做过类似的,给推荐下液相色谱条件,比如色谱柱、流动相等!!!先谢了!
最近在做对某主药中甲苯磺酸的含量测定,但是主药与该杂质的最大吸收波长不一致,所以我选的是杂质的波长,这样合理吗?在此波长下,对甲苯磺酸与主药的响应差10倍,那测定方法该用哪一种:外标法?







 我要推广仪器
我要推广仪器
 下载APP
下载APP