如题,有一反应产物,含甲醇、水、C4烯烃、二甲醚,用什么方法?什么柱子?可以一次进样做全分析?C4烯烃的异构体必须分离
乙二醇氧化制乙二醛反应,其中有好多副产物:甲醛(Formaldehyde,-19.5), 乙醇酸(glycolic,112), 乙醛(aldehyde, 20.8), 乙醛酸(glyoxalic acid, 111) 和 羟基乙酸(glycolic acid)。我查到有篇文章报道了这个反应产物的分析方法: 乙二醇(ethylene glycol, 197.3℃)和 乙二醛(glyoxal,51℃)用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](Reoplex400/Chromaton N column),甲醛(Formaldehyde,-19.5), 乙醇酸(glycolic,112), 乙醛(aldehyde, 20.8), 乙醛酸(glyoxalic acid, 111) 和 羟基乙酸用液相色谱(Agilent StableBond S-BC18 column)。由于水平有限,不能理解他为什么乙二醇和乙二醛要用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](所有产物均为液相),而其他的用液相呢?为什么这两种物质不直接用液相一块分析呢?
[color=#444444]乙二醇氧化制乙二醛反应,其中有好多副产物:甲醛(Formaldehyde,-19.5), 乙醇酸(glycolic,112), 乙醛(aldehyde, 20.8), 乙醛酸(glyoxalic acid, 111) 和 羟基乙酸(glycolic acid)。[/color][color=#444444]我查到2011年有篇文章报道了这个反应产物的分析方法:[/color][color=#444444] 乙二醇(ethylene glycol, 197.3℃)和 乙二醛(glyoxal,51℃)用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](Reoplex400/Chromaton N column),甲醛(Formaldehyde,-19.5), 乙醇酸(glycolic,112), 乙醛(aldehyde, 20.8), 乙醛酸(glyoxalic acid, 111) 和 羟基乙酸用液相色谱(Agilent StableBond S-BC18 column)。[/color][color=#444444]由于水平有限,不能理解他为什么乙二醇和乙二醛要用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](所有产物均为液相),而其他的用液相呢?为什么这两种物质不直接用液相一块分析呢?[/color]
甲醇及其产物的检测我用甲醇和水的混合溶液作为我的反应溶液,想要测定甲醇浓度的变化及产物的种类及浓度,请问各位学界长辈我该怎么做?选择气相可以么?
我要做乙醇水蒸气重整反应,液相产物有乙酸、乙醛、丙酮,想要测乙醇的转化率,需要找一个内标物,有人知道什么有机物合适吗?
马来酸二甲酯和苯甲醇在反应时颜色变黄,不知道是什么原因阿,有专家吗
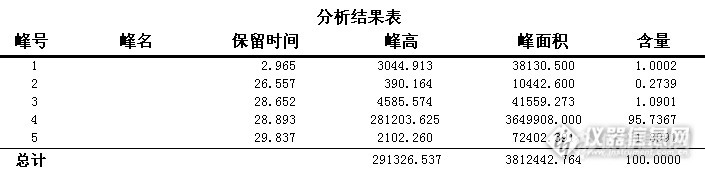
我想问一下怎么知道峰对应的是什么产物?甲醛,异丁醛,羟基新戊醛,1115酯,异丁醇,异辛醇,新戊二醇
用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
有机化学丁酮和醇钠反应的主产物并说明理由
我们这边[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]是Angilent7890,我做乙醇挥发性物质时,试剂配制用了乙醛,为什么乙醛的出峰时间和甲醇(乙醇里面的杂质)一样,是不是我的操作有问题啊?请高手解答,谢谢。[color=#DC143C]被测溶液:取无水甲醇50µ l与乙醛50µ l,加96%乙醇稀释至50.0ml。取该溶液100µ l,加96%乙醇稀释至10.0ml。[/color]色谱柱:熔融石英,长:30m,内径:0.32mm,固定相:聚[(氰丙基)(苯基)][二甲基]硅氧烷R (膜厚1.8µ m)。载气:色谱用氦气。线速度:35cm/s。检测器:氢焰离子化检测器。柱温:0-12min:40℃,12-32min:40→240℃,32-42min:240℃。进样口温度:200℃。检测器温度:280℃。进样量:1µ l。这是欧洲药典的要求,既然药典这样写的,甲醇和乙醛肯定能分开出峰的,但是我按这要求做过后,两个峰分不开。为什么?注:我用的仪器是Agilent [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]7890,但柱子是国产的,那是不是柱子的问题呢?
最近在做酯交换法合成碳酸二甲酯,碳酸丙烯酯跟甲醇反应,产物为丙二醇和碳酸二甲酯,定量分析不知如何分析。求助,仪器条件及内标物选定,谢谢啦!
按药典方法做,db-624色谱柱,0.25mm*1.4um,流速1ml,40度维持12分钟,每分钟10度升至240维持10分钟,分流5:1,出来的图如下,大神可否给看 看该怎么办?乙醛和无水甲醇峰都这么小吗?(图一)另一张是无水甲醇的(图二),如果不分流,无水甲醇和乙醇就连一起了(图三)求大神帮忙?[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251606373224_254_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251606376114_8345_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251606373624_9954_3443683_3.jpeg[/img]
做乙醇挥发性杂质测定时,按药典方法40度维持12分钟,每分钟10度升至240度,维持10分钟,使用db624柱子,0.25mm*1.4um,流速1ml,分流比5:1时,乙醛和无水甲醇峰特别小,峰形也不好(图一),单进无水甲醇的峰拖尾(图二),苯没出来,如果采取不分流进样,无水甲醇和乙醇峰就出一起了,大家做的图是什么样的,请大神帮忙看看怎么办?[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251637117241_6078_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251637118111_1179_3443683_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251637120861_5343_3443683_3.jpeg[/img]
液相色谱分析特例(一)—与甲醇反应的样品(上) 一客户拿来一样品,结构式见下,粗看也没什么特别,用户也没交代什么特殊情况,我想这种样品很简单,让学生帮我做,而且以前也做了同样的样品。化学结构式:[img=,207,149]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704032676_7363_1617661_3.png[/img]色谱条件:Agilent 1100 DAD检测器,自动进样器,Inertsil ODS-3 150*4.6mm,流速1.0ml/min,波长241nm,流动相甲醇:水=82:18,样品用甲醇溶解,进样量2ul。仪器平衡后,配好样品,启动程序自动运行,第一针还行,但基线不是很稳定,于是继续进样3针,好的话一般就没问题了。不过第二针出现四个峰,第三针出现较明显的三个峰,奇怪,这是什么原因,当第四针时跟第三针又没什么区别了。按理讲即使第一针不稳定,中间连续图谱也不应该出现多个色谱峰。我打开DAD看了不同图谱的光谱图,发现第一针和第四针的主峰的DAD图不一致,而第一图变化的主峰随着时间的延长不断减少,最终变成一个小杂质。[img=,351,221]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704034335_4399_1617661_3.png[/img][img=,379,236]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704033291_279_1617661_3.png[/img][img=,379,234]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704038257_3920_1617661_3.png[/img][img=,379,235]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704039929_8870_1617661_3.png[/img](四个光谱图)这些图谱明显显示,样品溶解后在不断的变化,其中最大可能是与甲醇发生了化学反应,随着反应的不断进行,最终变成了另外一个化合物,说实在的也非常有意思。分子上的三个醛基估计是反应的部位,从不同图谱的次序看,三个醛基都会反应,反应1-2个醛基是中间的不稳定态,最后三个醛基反应完,保留时间处于最后。样品又稳定了,如果谁用甲醇配,放置了一段时间(看上面实际时间大概1小时就ok了),再测,也非常好,而实际已经发生了巨变。与甲醇反应这么快,我极少遇到,而且是产生序列反应,一般能与流动相反应的样品大概在1-2%左右,因此分析者有时要注意,一些特特殊样品的特殊情况。下篇我们讨论改成乙腈作为流动相和溶剂的区别。[img=,211,168]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704038590_3596_1617661_3.png[/img][img=,218,171]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704039987_1878_1617661_3.png[/img][img=,204,159]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704044321_1260_1617661_3.png[/img][img=,228,174]https://ng1.17img.cn/bbsfiles/images/2024/03/202403181704043087_4531_1617661_3.png[/img](四个色谱图)。液相色谱分析特例(一)—与甲醇反应的样品(下)
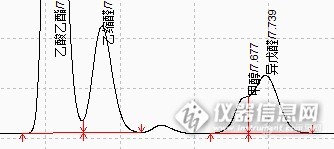
http://ng1.17img.cn/bbsfiles/images/2013/09/201309281119_468038_2451684_3.png乙缩醛和甲醇之间有一个峰,不知道是什么物质
实验目的:研究化合物合成反应监测及分离纯化一般步骤。一、监测合成反应TLC-MS检测,确定是否有合成产物:1、将待测样品通过硅胶板爬板分开。http://ng1.17img.cn/bbsfiles/images/2015/08/201508212000_562106_2307604_3.png(其实板爬的不歪,只是照片上有点歪,截完图就成这样了。)2、利用TLC-MS仪器快速检测TLC板的两个点,确定目标物。http://ng1.17img.cn/bbsfiles/images/2015/08/201508212002_562107_2307604_3.png点1质谱检测图谱:http://ng1.17img.cn/bbsfiles/images/2015/08/201508212106_562110_2307604_3.png点2质谱检测图谱:http://ng1.17img.cn/bbsfiles/images/2015/08/201508212111_562115_2307604_3.png结论:经TLC-MS检测,合成反应有效,点2中含有分子量432的目标物,点1为原料。二:液相分析由于目标物极性较小,液相分析色谱柱选用C18色谱柱保留太强,因此选用保留稍弱的C8色谱柱。色谱条件如下:色谱柱: C8 5 μm 100 Å 4.6*250 mm流动相:A:水 B:乙醇流速:1.0 mL/min检测器:ELSD,65℃梯度:TimeB%0752510035100原样分析图谱:http://ng1.17img.cn/bbsfiles/images/2015/08/201508212115_562117_2307604_3.png27.448min峰为目标峰三、分离纯化:经测试:硅胶柱纯化条件不能把目标物前21-26 min杂质分离除去,C8柱纯化条件不能将26.1 min和27.9 min杂质分离除去。因此最终方案选用C18色谱柱,以甲醇和二氯甲烷为流动相,达到了很好的分离效果。纯化条件如下: 色谱柱:C18 10 μm 100 Å 30*250 mm 流动相:A:甲醇 B:二氯甲烷 流速:35 mL/min 紫外波长:210 nm(红色信号线) ELSD:65℃(浅蓝色信号线) 梯度:TimeB%053035 进样量:300 mg(10mL甲醇溶解) 分流: 流动相进紫外检测器与蒸发光散射检测器的分流比为34.5:0.5 制备图谱如下:http://ng1.17img.cn/bbsfiles/images/2015/08/201508212116_562118_2307604_3.png馏分收集:收集10.3-11.7 min和21.3-23.4 min馏分四、纯度检测(条件为液相分析条件):1、 10.3-11.7 min馏分:取200 uL馏分,氮吹干,加200 uL甲醇溶解,进样10 uL检测,检测结果如下:http://ng1.17img.cn/bbsfiles/images/2015/08/201508212117_562119_2307604_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508212118_562120_2307604_3.png2、 21.3-23.4 min馏分:取200 uL馏分,氮吹干,加100 uL甲醇溶解,进样10 uL检测,检测结果如下:http://ng1.17img.cn/bbsfiles/images/2015/08/201508212119_562121_2307604_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508212119_562122_2307604_3.png结论:1、合成反应可以利用TLC-MS设备快速监测合成反应成功与否。2、根据待分离样品的极性和硅胶板上的保留,选择合适的填料和流动相,摸索纯化条件。五、总结:1、合成反应可以利用TLC-MS设备快速监测合成反应成功与否。2、根据待分离样品的极性和硅胶板上的保留,选择合适的填料和流动相,摸索纯化条件。六、实验心得:1、利用TLC-MS检测仪,直接检测TLC板上的样品点,不需要将硅胶板上的点刮下来再处理后扫质谱。2、部分极性较小的样品可以用C18色谱柱,配二氯甲烷等弱极性试剂作为流动相进行分析或纯化。3、对于紫外吸收弱的样品,可并联蒸发光散射检测器,由于ELSD为分析型检测器,在大流速制备情况下,需要调节分流比,ELSD的分流速不能高于0.5 mL/min,以防检测器过载。4、制备条件下,由于并联检测器分流比差别大,且检测过程耗时不同,两个检测器在图谱中出现信号的时间就会有差异。这时需要提前判断两个信号的相对延迟时间,通常以流速大的紫外信号作为收集信号。5、紫外下吸收很弱的样品,放大到制备,加大上样量后,紫外下也会有吸收,也可作为制备收集的信号。6、当制备馏分溶剂为弱极性溶剂时,而检测条件的流动相为较强极性流动相,或存在溶剂不互溶问题,需要将制备馏分浓缩干,再用接近检测条件流动相极性的溶剂溶解,进行分析,避免溶剂效应。
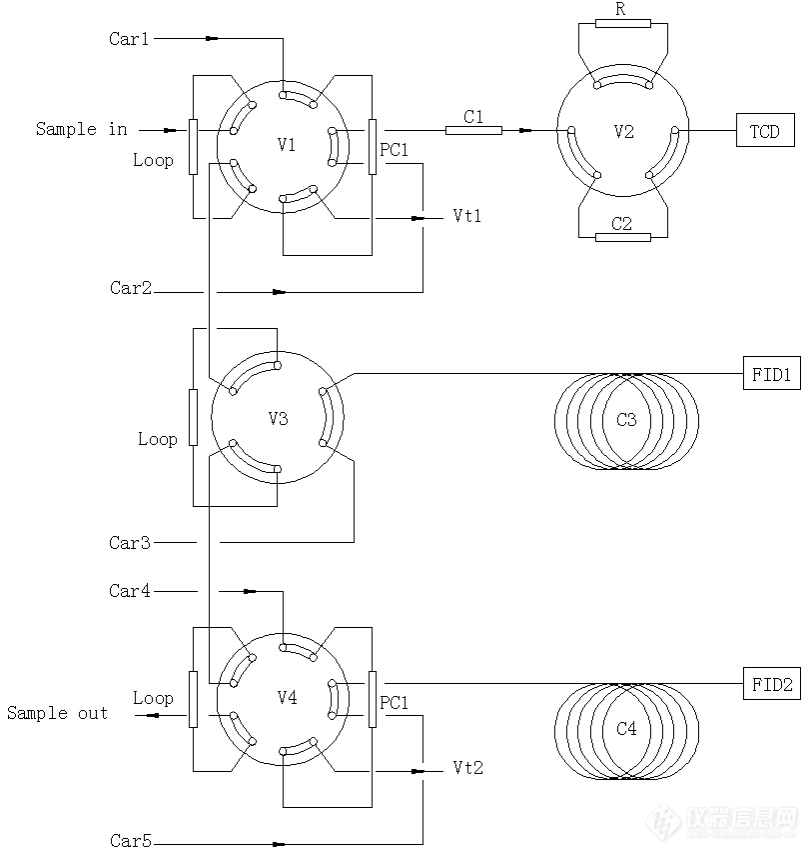
[align=center][size=24px]甲醇制取烯烃产物分析系统原理介绍[/size][/align][align=center][color=black]概述[/color][/align][color=black]使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]配置三检测器四切换阀系统,测定甲醇制取烯烃反应过程中的各种气体组分含量。[/color][align=center][color=black]一 背景介绍[/color][/align][color=black]乙烯、丙烯等低碳烯烃是重要的基本化工原料,随着我国国民经济的发展,特别是现代化学工业的发展对低碳烯烃的需求日渐攀升,供需矛盾也将日益突出。甲醇制乙烯、丙烯的MTO工艺和甲醇制丙烯的MTP工艺是重要的化工技术。该技术以煤或天然气合成的甲醇为原料,生产低碳烯烃,是发展非石油资源生产乙烯、丙烯等产品的核心技术。[/color][color=black]甲醇制取烯烃的反应过程中各工段的产物组成较为复杂,包括甲醇、二甲醚、小分子烷烃烯烃类、以及少量二氧化碳和永久气体等组分,使用简单的单根[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱难以实现分离。如果采用多次进样的方法,无疑分析效率会显著降低。那么设计可单次进样,可在线连接的专用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析系统会极大提高分析效率。[/color][align=center][color=black]二 系统结构原理[/color][/align][color=black]本例采用Shimadzu 的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]GC-2014,设计甲醇制取烯烃过程气体分析系统,系统结构原理如图1所示,系统中含有三检测器——两个FID检测器、一个TCD检测器——四支自动阀,具有并行的三路分析通道。[/color][color=black]通道1采用十通阀进样反吹并辅助以六通阀切换色谱柱的方法,用以测定样品中的少量氢气、氧气、氮气、一氧化碳、二氧化碳等组分含量。[/color][color=black]通道2采用六通阀直接进样、PLOT Q毛细管柱分离的方法,用以测定样品中甲醇和二甲醚等组分的含量。[/color][color=black]通道3采用十通阀进样反吹、氧化铝毛细管柱分离的方法,用以测定样品中的丙烯、乙烯以及其他烃类化合物含量。[/color][color=black]本系统可以实现一次进样完成样品所有组分的分离测定,并且可以实现在线或者离线方式的采样。[/color][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091651539518_1703_1604036_3.jpg[/img][/align][align=center]图1 甲醇制取烯烃产物分析系统原理图[/align][align=center][color=black]三 工作流程讲解[/color][/align][align=center][color=black]本分析系统的工作过程简述:[/color][/align][align=center][color=black]通道1的工作过程:[/color][/align][color=black]1 取样[/color][color=black]如图1所示,此时将样品通入定量环(样品流经 sample in - loop -sample out)。[/color][color=black]2 进样[/color][color=black]系统启动数据采集的瞬间,十通阀V1旋转36度,此时样品被载气携带进入预分离色谱柱PC1中(样品流经 car1 - loop - PC1 - C1 - C2 -TCD1 )。[/color][color=black]样品在PC1中被预分离,其中较轻的组分(氢气、氧气、氮气、甲烷、一氧化碳)作为合峰流入C2色谱柱。[/color][color=black]3 反吹[/color][color=black]当样品中的二氧化碳之前的组分全部流入色谱柱C1之后,十通阀V1旋转36度,此时预分离色谱柱PC1中的载气流速反方向流动,保留时间较长的重组分被反吹流出PC1柱(样品流经 car1 - PC1 - Vent1)。[/color][color=black]4 色谱柱选择[/color][color=black]样品在C1色谱柱中被分成两部分,一部分为氢气、氧气、氮气、甲烷、一氧化碳的合峰,另一部分为二氧化碳和其他烃类。[/color][color=black]当合峰完全流入色谱柱C2中时,V2阀旋转60度,合峰中的氮气、一氧化碳、甲烷等组分被封闭在色谱柱C2中。C1色谱柱中的二氧化碳流出色谱柱,由于色谱柱保留时间配合的关系,氢气和氧气也会流出进入TCD检测器。此时观察到的TCD出峰顺序为氢气、氧气、二氧化碳。[/color][color=black]5 复位[/color][color=black]当乙炔完全流出色谱柱C1之后,V2阀旋转60度,恢复到系统的初始状态,C2中封闭的组分,再次流出并在TCD1上出峰,其顺序为氧气、氮气、甲烷、一氧化碳。[/color][align=center][color=black]通道2 的工作过程:[/color][/align][color=black]1 取样[/color][color=black]如图1所示,此时将样品通入定量环(样品流经 sample in - loop -sample out)。[/color][color=black]2 进样[/color][color=black]系统启动数据采集的瞬间,六通阀V3旋转60度,此时样品被载气携带进入色谱柱C3中(样品流经 car3 - C3 - FID1)。[/color][color=black]色谱柱C3为PLOT Q毛细管柱,可以将样品中的甲醇和二甲醚分离开,此通道的管路、阀和定量环需要特殊处理,予以保温和进行惰性化处理。[/color][align=center][color=black]通道3 的工作过程:[/color][/align][color=black]1 取样[/color][color=black]如图1所示,此时将样品通入定量环(样品流经 sample in - loop -sample out)。[/color][color=black]2 进样[/color][color=black]系统启动数据采集的瞬间,十通阀V4旋转36度,此时样品被载气携带进入预分离色谱柱PC2中(样品流经 car3 - loop -PC3 - C3 - TCD2)。[/color][color=black]样品在预分离色谱柱PC2(采用了强极性色谱柱)中分离为较轻组分(烃类物质永久气体)和较重组分(极性较强组分包括甲醇和二甲醚)。[/color][color=black]其中保留较弱的烃类组分流入色谱柱C4(氧化铝毛细管柱),并在FID2检测器上被检测到。[/color][color=black]3 反吹[/color][color=black]当色谱柱PC2中的较轻组分完全流入色谱柱C4中,十通阀V4再次旋转36度,此时色谱柱PC2内部的载气反向流动,将保留时间较强的组分反吹流出系统。[/color][color=black]最终总系统复位,准备下次进样。[/color]系统的典型谱图如图3所示:[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091651542232_7794_1604036_3.jpg[/img][/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091651544849_9664_1604036_3.jpg[/img][/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111091651544537_9697_1604036_3.jpg[/img][/align][align=center]图3 系统典型谱图[/align]
我们这边[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]是Angilent7890,我做乙醇挥发性物质时,试剂配制用了乙醛,为什么乙醛的出峰时间和甲醇(乙醇里面的杂质)一样,是不是我的操作有问题啊?请高手解答,谢谢。被测溶液:取无水甲醇50µ l与乙醛50µ l,加96%乙醇稀释至50.0ml。取该溶液100µ l,加96%乙醇稀释至10.0ml。色谱柱:熔融石英,长:30m,内径:0.32mm,固定相:聚[(氰丙基)(苯基)][二甲基]硅氧烷R (膜厚1.8µ m)。载气:色谱用氦气。线速度:35cm/s。检测器:氢焰离子化检测器。柱温:0-12min:40℃,12-32min:40→240℃,32-42min:240℃。进样口温度:200℃。检测器温度:280℃。进样量:1µ l。这是欧洲药典的要求,既然药典这样写的,甲醇和乙醛肯定能分开出峰的,但是我按这要求做过后,两个峰分不开。为什么?注:我用的仪器是Agilent [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]7890,但柱子是国产的,那是不是柱子的问题呢?
我现在需要分析乙醇脱氢反应的气相产物,将气相产物进色谱柱PQ柱,柱温程序是150℃恒温25分钟,进样口温度150,TCD检测器温度为150,气相产物里面主要含有反应保护气氮气、产物乙醛、乙醇。出峰的情况是:氮气峰很好,但在氮气之后鼓了一个大包,这是为什么呢
各位专家好!我现在使用一台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],分析乙醇和水反应后的产物。用PorapakQ分析反应后的乙醇和水,Tdx分析CO,CO2,C2H4,H2作载气。产物中还有H2。我想通过计算分析出产物中各组分含量。两根柱子无法统一计算,请各位专家指导!
最近在检测氨基乙醛缩二甲醇这个产品,用一个滴定的方法,颜色不好判断,哪位高手指点一下,最好有什么先进的方法,比如GC。
分析白酒时乙醛和甲醇时间挨的近,出了乙醛峰很好,但是甲醇就不行了,又矮又长,怎么办啊?调节什么可以[em09501]
有个检测正丁基锂浓度的方法,其中向正丁基锂中加入1,2-二溴乙烷进行反应。不知反应后,产物是什么?
BF3- 甲醇与ROOH反应甲基化,生成的产物应该是ROOCH3,还是ROOBF3呢?我觉得是前者,别人说是后者。
最近用DMSO作溶剂,检测某草酸盐中的残留溶剂丙酮、异丙醇等,加了草酸盐之后,发现在丙酮和异丙醇之间产生一个未知峰,但是空白中是没有的。以前做某盐酸盐时,也是用的DMSO为溶剂,也是DB-624的柱子,也是顶空进样,结果也是在丙酮和异丙醇之间有一个未知峰,后来气质定性为二甲基硫醚,是盐酸和DMSO反应生成的。综合两次,考虑应该是DMSO与酸反应生成了新物质即二甲基硫醚或者是其他物质。但是我对化工合成不是很了解,希望前辈们指点。期待大家的帮助。网上查到二甲基亚砜的工业合成工艺:采用甲醇与二硫化碳为原料合成二甲基硫醚,温度380度,常压,同时可副产10%以上的甲硫醇等副产品。二甲基硫醚生产二甲基亚砜采用氧化和精制两个工序,在氧化工序中,二甲基硫醚与二氧化氮(NO2)在液相中反应生成二甲基亚砜,同时二氧化氮被还原成氧化氮,采用逆流塔式液相氧化反应器,塔底温度60度,塔顶温度25度。二氧化氮由亚硝酸钠和浓硫酸反应制备,被还原后生成的NO用氧气氧化重新制得二氧化氮。未反应的二甲基硫醚经闪蒸从产物中分离,再循环使用,二甲基亚砜粗产品用氢氧化钠进行中和处理。 二甲基亚砜精馏采用三塔流程,经蒸发脱盐、减压精馏脱水和真空精馏获得纯度为99%以上的精制二甲基亚砜产品。
近来做一个反应,反应产物中可能会有CO、H2、O2 、CO2、CH4、H2O、甲烷、乙烯、环氧乙烷、乙醇、乙醛、乙酸、乙醚等。我想知道产物的组成,手头上有一台岛津GC2014,RTX-1和RTX-5色谱柱各一根。请问专家和楼主们: (1)一台色谱和两根色谱柱能分析产物中所有的组分吗?是否还需要一台色谱?另外,还需要购买哪种色谱柱? (2)分析时,同一台色谱上安装的两根色谱柱采用并联还是串联,哪种安装和操作起来更容易些? 急等!谢谢!
我用的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]器是安捷伦的7820A型,主要是是做无水乙醇挥发杂质的分析。 刚用这根柱子做的时候,乙醛和无水甲醇峰的分离度能达到2左右!(药典规定大于1.5)后来对仪器啊,色谱柱都不怎么熟悉,就又用这柱子做了一种有机胺化合物的含量。结果后来再做无水乙醇的时候,两个峰之间分离度就不够了,换了根新柱子也不行。而且现在的甲醇峰还有拖尾的现象,不知道是怎么回事。色谱柱:DB-624毛细管柱分离度:20:1升温程序: 时间 min 温度 ℃ 色谱柱 0---12 40 12---32 40到240 32---42 240 进样部位 -- 200 检测器 -- 280
怎么分开甲醇和乙醛、氯乙烯色谱峰?我用SE-30(FID)分不开这几种物质?
我们在做药用乙醇的挥发性杂质分析,遇到了问题,甲醇和乙醛的分离度不好,甲醇的峰也容易拖尾,将分流比调大是可以达到分离度的要求,但是又达不到检出限了,请大侠们帮帮忙,那些条件应该怎么设置
求助。。请问各位大佬,我做了个催化反应,溶剂是水,产物中有甲醇,环己醇等等,进[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的时候我是用的乙酸乙酯萃取,其余产物可以萃取出来,但是甲醇萃取不出,不能定量;若用乙醇稀释下反应产物,很多物质又不会出峰;怎样才能在色谱中准确测出甲醇含量?(产物:甲醇,环己醇,苯酚,2-甲氧基环己醇,环己酮)







 我要推广仪器
我要推广仪器
 下载APP
下载APP