想请教十水硫酸钠含量测定(杂质有:硼酸钠\对甲苯磺酸),我想用钡盐沉淀重量法,但怕杂质干扰,也想用强阳离子树脂交换法将钠交换出来后用氢氧化钠滴定,但也怕杂质干扰,不知还有什么方法检测,请大家不怜赐教!
哪位同道有 对氨基二甲基苯胺比色法 GB 18056 –2000 附录A 用来检测甲硫醇的,急需!!!
有哪位大侠做过三(三甲基硅烷)硼酸酯或三(三甲基硅烷)磷酸酯的分析啊?用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的话选用何种柱子合适啊?
求助哪个朋友有双氧水甲酸硼酸硫酸乙醇/乙二醇丁谜 硫酸亚锡的国家标准啊 求哪朋友有这样的 急求~~谢谢的小弟QQ112277861
异硫氰酸苯酯做为衍生试剂时,可以用分析纯的替代吗?我们刚要分析一种新的氨基酸,需要用异硫氰酸苯酯,可是到处都买不到色谱纯的,请问大家,可以用分析纯的代替吗??多谢多谢了.[em0808]
在一个牛奶香精中分析出了甲基硫代磺酸甲酯(CAS:2949-92-0),也在GB2760里。我在想这个原料会不会是二甲基二硫醚氧化过来的?请各位大神解答!谢谢
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
求助关于四甲基频哪醇重排副反应的文献!
对氨基二甲基苯胺光度法测定水样中硫化物的几个问题,希望那个大侠帮忙解决一下?我们公司使用的工艺是低温甲醇洗,测定的是其贫甲醇中的微量硫化氢含量?在测定过程中遇到的问题是:1、回收率达不到95%以上;2、用不同的稀释倍数测定同一个样品,其测定结果相差很大。3、贫甲醇中的硫化氢含量其实不多,但是把上述方法中药品加进去后,其颜色一直很深,且最后不会变成蓝色,但是用硝酸银定性,又感觉硫化氢很多。
有哪位大侠做过甲基硼酸的测试啊?谢谢指教!
四苯硼酸钠和四苯硼钠是同一种试剂吗?
请问如何测定水样中二甲基二硫代氨基甲酸钠(福美钠)的浓度?请高手指教最好详细一点,谢谢。
[size=4][font=宋体]求香菇上氟乐灵,甲基毒死蜱,毒死蜱,氯杀螨,乙硫磷,氯苯嘧啶醇[/font][/size][size=4][font=Times New Roman] [/font][/size][size=4][font=宋体]氟虫腈的前处理方法,能够有效去除杂质的,谢谢大家,这个香菇上的杂质太多了,用弗洛里硅土不管用,杂质很多。[/font][/size]
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
薄层色谱法l 应用范围本方法适用于化妆品中丙酸、硼酸的检验。2原理检样经浸提后,置于薄层板上,利用不同组分在展开剂中溶解度和与层析板的吸附剂的亲合力不同而分离,显色剂显色后加以鉴别。3试剂3.1 展开液:称取11.5g碳酸铵于200ml容量瓶中,加入0.6mol/L NH4OH到刻度。此溶液含NH,约1.02%(W/V),取120ml此溶液加到240m1 95%乙醇和40ml 正丙醇中。3.2 显色剂A:称取1g 2-氯4-硝基苯二叠氮(2-chlor-4-nitro-benzenediazouium natha-lene-2-sulonate)于烧杯中,加入100m1水,配成1%溶液,用中速滤纸过滤。使用前配制。显色剂B:称取2g葡萄酸,溶于20ml 水中,转移至100ml容量瓶,加入2ml香草醛和20m1 95%乙醇,加正丁醇至刻度。3.3 丙酸标准溶液:称取相当含丙酸0.4g的丙酸或丙酸盐,溶于20ml水中,转移至100ml容量瓶,加水稀释至刻度。此溶液含丙酸0.4%(1)。3.4 硼酸标准溶液:称取0.6g硼酸溶于20ml水中,转移至100ml容量瓶,加水稀释至刻度。此溶液含硼酸0.6%。4仪器4.1高效纤维素薄层板: Merck l5035或Merck l5036,或等效物。4.2层析缸:玻璃标本缸,带盖。5 分析步骤5.1样品前处理称取约2.0g样品,加入10ml 95%乙醇 水(1 1),在超声波匀浆器内匀浆15min,在500r/min下离心10min,取上清液为待测溶液。5.2 分别取2μ1硼酸标准液、丙酸标准液及检液点样于薄层板的点样区,每次1μ1。将薄层板置于内装有20ml展开液的层析缸内,盖上缸盖,进行层析(2)。5.3 展开液前沿移动6cm以后,取出薄层板自然风干。5.4 将显色剂A喷布在薄层板上。自然风干1min后,将显色剂B喷布于其上。将薄层板置于100℃烘箱加热15min。取出烘箱,检查检样之色斑并与标准进行比较(3),以鉴别是否存在硼酸(盐)或丙酸/丙酸盐,并可估测其含量是否接近或超过最大允许含量(1)。
测定大气中多环芳烃时,利用内标六甲基苯,打算用异辛烷配制,但不知道其在异辛烷中的溶解度或者饱和度,哪问高手知道指教一下?有没有做过的,讲授一下在配备过程中的注意事项。多谢啦
常用试剂的配制作业规范5.22.1 1%酚酞 5.22.1.1 所需药品:酚酞5.22.1.2 用途:酸碱指示剂5.22.1.3 配制方法:称取1g酚酞,用100mL无水乙醇溶解5.22.2 0.1%甲基红5.22.2.1 所需药品:甲基红5.22.2.2 用途:配制甲基红-溴甲酚绿混合指示剂5.22.2.3 配制方法:称取1g甲基红,用1000mL无水乙醇溶解5.22.3 0.1%溴甲酚绿5.22.3.1 所需药品:溴甲酚绿5.22.3.2 用途:配制甲基红-溴甲酚绿混合指示剂5.22.3.3 配制方法:称取1g溴甲酚绿,用1000mL无水乙醇溶解5.22.4 甲基红-溴甲酚绿混合指示剂5.22.4.1 用途:测蛋白质用指示剂5.22.4.3 配制方法:临用时按0.1%甲基红:0.1%溴甲酚绿=1:5体积比混合而成5.22.5 1%淀粉指示剂5.22.5.1 所需药品:可溶性淀粉5.22.5.2 用途:活性氯测定指示剂5.22.5.3 配制方法:称取1.0g可溶性淀粉,加少量RO水搅匀,随后一面搅拌一面加入热水约60mL再将此溶液煮沸2-3min,静置冷却,加NaCl20g,溶解后再加RO水至100mL(可冷藏备用)5.22.6 3%硼酸溶液5.22.6.1 所需药品:硼酸5.22.6.2 用途:测定蛋白质5.22.6.3 配制方法:称取3.0g硼酸,用RO水溶解并定溶至100mL5.22.7 40%NaOH溶液5.22.7 所需药品:NaOH5.22.7.2 用途:测定蛋白质5.22.7.3 配制方法:称取416.7g NaOH,溶解于583.3g RO水中5.22.8 邻联甲苯胺溶液5.22.8.1 所需药品:分析纯Hcl、邻联甲苯胺5.22.8.2 用途:检测水中余氯5.22.8.3 配制方法:量取150mL浓盐酸用RO水稀释至500mL。称取1.00g邻联甲苯胺(或1.35g邻联甲苯胺盐酸盐),并吸取5mL配制好的盐酸一同放入玻璃研钵器中研成糊状。然后置入1000mL的容器中加150mL蒸馏水稀释,同时加入495mL稀盐酸并用玻璃棒充分搅拌,最后用蒸馏水稀释至1000mL。5.22.9 0.1mol/L NaOH标准溶液5.22.9.1 配制:称取4.17gNaOH(纯度96%),加蒸馏水溶解,并稀释至1000mL。5.22.9.2 标定:精确称取0.2g在105-110℃烘至恒重的基准邻苯二甲酸氢钾,溶于50mL新煮沸过的冷水中,加2滴酚酞指标剂,用配制好的NaOH溶液滴定至溶液呈粉红色。5.22.9.3 同时作空白试验。5.22.9.4 结果表示C(NaOH)= 式中:C(NaOH):氢氧化钠标准溶液之物质的量浓度,mol/Lm:邻苯二甲酸氢钾之质量,gV1:氢氧化钠溶液之用量,mLV2:空白试验氢氧化钠溶液之用量,mL0.2042:与1.00mL NaOH标准溶液[C(NaOH)=1.000mol/L]相当的基准邻苯二甲酸氢钾的质量(g)5.22.10 0.1mol/L HCl标准溶液5.22.10.1 配制量取9mLHCl,加适量水并稀释至1000mL。5.22.10.2 标定准确称取0.15g在270-300℃干燥到恒重的基准无水碳酸钠,加50mL水使之溶解,加10滴溴甲酚绿甲基红混合指示剂,用配制好的HCl 溶液滴定至溶液由绿色转变为紫红色,煮沸2min,冷却至室温,继续滴定至溶液由绿色变为暗紫色。5.22.10.3 同时做空白试验。5.22.10.4 结果表示C(HCl)= 式中:C(HCl):盐酸标准溶液之物质的量浓度,mol/Lm:无水碳酸钠之质量,gV1:HCl溶液之用量,mLV2:空白试验HCl溶液之用量,mL0.0530:与1.00mL HCl标准溶液[C(HCl)=1.000mol/L]相当的基准无水碳酸钠的质量(g)5.22.11 0.1mol/L Na2S2O3标准溶液5.22.11.1 配制:称取26克硫代硫酸钠及0.2克碳酸钠,加入适量新煮沸过的冷水使之溶解,并稀释至1000mL,混匀,过滤后备用。5.22.11.2 标定准确称取约0.15克在120℃干燥至恒量的基准重铬酸钾,置于500mL碘量瓶中,加入50mL水使之溶解。加入2g碘化钾,轻轻振摇使之溶解。再加入20mL硫酸(1+8),密塞,摇匀,放置暗处10min后用250mL水稀释。用硫代硫酸钠标准溶液滴至呈浅黄绿色,再加入3mL淀粉指示液(0.5%),继续滴定至蓝色消失而显亮绿色,反应液及稀释用水的温度不应高于20℃5.22.11.3 同时做空白试验5.22.11.4 结果表示C(Na2S2O3)= 式中:C(Na2S2O3): Na2S2O3标准溶液之物质的量浓度,mol/Lm:无水重铬酸钾之质量,gV1:Na2S2O3溶液之用量,mLV2:空白试验Na2S2O3溶液之用量,mL0.04903:与1.00mL Na2S2O3标准溶液[C(Na2S2O3)=1.000mol/L]相当的基准重铬酸钾的质量(g)5.22.11.5 0.005mol/L、0.01mol/L Na2S2O3标准溶液临用前取0.1mol/L Na2S2O3标准溶液,加新煮沸过的冷水稀释制成。5.22.12 各种试剂的配制和标定周期:5.22.12.1 配制周期至少1次/月。5.22.12.2 标定周期至少1次/月。5.22.13 常用酸碱浓度(市售)试剂名称 分子量 含量%(m/m) 相对密度 浓度(mol/L) 冰乙酸 60.05 99.5 1.05 17(CH3COOH) 盐酸 36.45 36-38 1.18 12(HCl) 硫酸 98.1 96-98 1.84 18(H2SO4)
我国对化肥中钾含量的测定以四苯硼酸钾重量法应用最广,该方法具有测定结果准确的特点。 钾是植物营养三要素之一,它与氮、磷元素不同,主要呈离子状态存在于作物细胞的汁液中,具有高度的渗透性、流动性和再利用的特点。化肥中的钾元素能促使作物生长健壮,茎秆粗硬,增强对病虫害和倒伏的抵抗能力,促进糖分和淀粉的生成,从而使农作物增产,提高农产品品质。目前,我国对化肥中钾含量的测定以四苯硼酸钾重量法应用最广,该方法具有测定结果准确的特点,但耗时较长。下面笔者将以复混肥料(复合肥料)为例,结合实际检验过程中的一些问题,就该方法的原理、方法及注意事项等进行阐述,不妥之处请同行批评指正。 测定原理 在弱碱性介质中,以四苯硼酸钠溶液为沉淀剂沉淀试样溶液中的钾离子,生成白色的四苯硼酸钾沉淀,将沉淀过滤、洗涤、干燥、称重。根据沉淀质量计算化肥中钾含量。反应式为: K++Na →K ↓+ Na+ 操作步骤 1.试样溶液的制备 称取试样(按GB/T8571规定所制备的样品)约2g-5g(含氧化钾约400mg),精确至0.0002g,置于250mL锥形瓶中,加水约150mL,加热煮沸30min,冷却,定量转移到250mL容量瓶中,用水稀释至刻度,混匀,干过滤,弃去最初滤液50mL。 2.试液处理 吸取上述滤液25mL于250mL烧杯中,加EDTA溶液 (40g/L)20mL(含阳离子较多时可加40mL),加2-3滴酚酞指示剂(5g/L乙醇溶液),滴加氢氧化钠溶液(400g/L)至刚出现红色时,再过量1mL,盖上表面皿,在良好的通风橱内缓慢加热煮沸15min,然后冷却,若红色消失,再用氢氧化钠(400g/L)调至红色。(如果试样中含有氰氨基化物或有机物时,在加入EDTA溶液之前,先加溴水和活性炭处理:加入5%的溴水溶液5mL,将该溶液煮沸脱色至无溴颜色为止,若含其他颜色,将溶液体积蒸发至小于100mL,冷却后加0.5g活性炭充分搅拌使之吸附,然后过滤、洗涤,洗涤时每次用水约5 mL,次数为3-5次,并收集全部滤液)。 3.沉淀及过滤 在不断搅拌下,于盛有试样溶液的烧杯中逐滴加入四苯硼酸钠沉淀剂(15g/L),加入量为每含1mg氧化钾加沉淀剂0.5mL,并过量7mL,继续搅拌1min,静置15min以上,用倾滤法将沉淀过滤于预先在120℃下恒重的4号玻璃坩埚式滤器内,用四苯硼酸钠洗涤液(1.5g/L)洗涤沉淀5-7次,每次用量约5mL,最后用水洗涤2次,每次用量约5mL。 4.干燥 将盛有沉淀的坩埚置于120℃±5℃干燥箱中,干燥1.5h,取出后置于干燥器内冷却,称重。 5.同时做空白试验(除不加试液外,分析步骤及试剂用量同上述步骤)。
[em09509]硫代硫酸钠滴定液配制时,加硫代硫酸钠+0.2g碳酸钠,加水,煮沸,放置2周后过滤。为什么要放置2周呢?是为了让它沉淀后,上层清液便于过滤吗?以前做K的测定是,配制那个四苯硼酸钠溶液,也让放置一夜,再过滤,是不是也是让它充分沉淀,仅过滤上层清液,便于过滤?
GB 23487-2009 食品添加剂2-甲基-3-呋喃硫醇[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=174134]GB 23487-2009 食品添加剂 2-甲基-3-呋喃硫醇.pdf[/url]
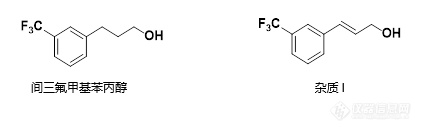
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
要做某样品中的二氯对甲基苯甲酸的残余。该物质沸点330.51.首选了ffap柱二氯对甲基苯甲酸标品用溶剂溶解,不出峰。衍生化,出很小的峰。2.rtx-1柱这么高的沸点,6min出峰了。







 我要推广仪器
我要推广仪器
 下载APP
下载APP