[color=#444444]请问在分子筛B酸位上形成的甲氧基中的甲基,和乙酰基中的甲基,两者的红外吸收峰有何不同?就是波数怎么变化,哪个高哪个低?非常感谢![/color]
我们想用GC来检测乙酰氧基乙酰氯,因为原来是采用滴定方式,过程比较烦琐,而且结果准确度不高。
由于手上没有2,3,4-三甲氧基乙酰苯(2,3,4,-trimethoxyacetophenone,C11H14O4)的对照品,所以想求助大家有没有它的光谱图,有的请发一份上来好吗?谢谢大家帮忙啦,
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
求大神请教指教,我是安捷伦[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]7890A,FPD检测器,新的不分流玻璃衬管,HP-5柱子才换几个月,初始温度100,保持2分钟,速率8,陡升到250度,保持1分钟(以前按照这条件做的)!配的含0.2ug/ml的乙酰甲胺磷同含0.36的氧乐果的混标,其他化合物都出峰了,但是这两个还是不出峰,分别用10浓度上机,出峰了,但是是拖尾,后来我又切掉检测器端柱子几十厘米,换了进样针、螺母同进样处的密封圈圈还是没改变,请问需要怎么做呢?谢谢
螺内酯片属于那类药物?我们用液相检测其含量和含量均匀度,每次都会无缘无故出现异常峰面积,特别头疼,希望大神说出自己的看法~~
第一步:甲氧基聚乙二醇的合成聚乙二醇在无水二氯甲烷中与金属钠作用生成聚乙二醇钠, 然后与碘甲烷反应即得。一甲氧基聚乙二醉、双端都反应的二一甲氧基聚乙止醇和未反应的聚乙二醇的反应混合物硅胶柱层析色潜提纯可以得到纯净的甲氧基聚乙二醇第二步:甲氧基聚乙二醇丁二酸单醋的合成将甲氧基聚乙二醇(Me-PEG-2000)、丁二酸酐和催化剂加入盛有二氯甲烷的圆底烧瓶中, 磁力搅拌使固体完全溶解后, 室温搅拌反应过夜。反应液分别用盐酸水溶液、氢氧化钠水溶液和甲醇水溶液依次洗涤。有机相经无水MgSO4干燥, 过滤除去干燥剂, 减压蒸除有机溶剂, 残留物以石油醚结晶, 收率90%。第三步:甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺的合成甲氧基聚乙二醇丁二酸单酷先经N一羟基丁二酰亚胺(NHS)活化, 然后缓慢滴加人到二硬脂酰磷脂酰乙醇胺(DSPE)的三氯甲烷中, 加料完毕后继续反应4h, 蒸除溶剂, 浓缩液在乙醚中结晶,硅胶柱层析色谱提纯可以得到自色粉末状固体的。甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺。来源:中国标准物质网
氧化乐果、乙酰甲胺磷用哪种基质配制,响应值高?这两种农药用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]、[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]来做,都存在吸附现象,出峰很小,用基质配制,出峰会高些,用哪种蔬菜基质,峰会大些?
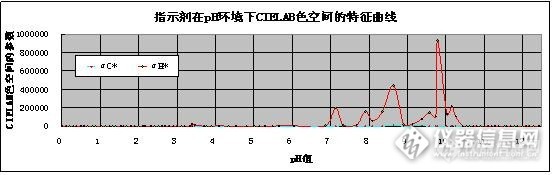
新技术6:乙氧基黄叱精指示剂CIELAB色空间颜色特征摘要:用CIE 1976(L*,a*,b*)色空间方法对乙氧基黄叱精在不同pH环境进行了测量,发现其变色点有8个,与文献记载有较大差异。用于指示的指标a*值、b*值、C*值、△E1、△E1-V可以作为变色的指标,发现在其在高pH环境有新的颜色变化,拟补了传统资料的不足。关键词:乙氧基黄叱精,CIE,色度值,数字化 前言 乙氧基黄叱精(Ethoxychrysoidine Hydrochloride)是常用指示剂,分子式C14H16N4O·HCl,分子量292.77,外观为为深红棕色或黑褐色粉末,在水或乙醇中溶解。pH变色域:pH3.5~ 5.5(红→黄)。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669935_1722582_3.jpghttp://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif图1. 乙氧基黄叱精的化学结构式 采用CIELAB色空间方法研究乙氧基黄叱精指示剂在不同pH溶液中变色现象的文献未见报道。用色空间方法首次测定了乙氧基黄叱精指示剂的L*、a*、b*等色度值参数,与pH值的对应关系,绘制出乙氧基黄叱精指示剂变色的L*a*b*色空间色度学参数与pH值的关系图,为精确描述乙氧基黄叱精指示剂变色特征奠定了基础。1. 实验部分1.1试剂、仪器与测量条件 0.5mol/L H2SO4溶液,0.5 mol/L NaOH溶液,乙氧基黄叱精溶液(取乙氧基黄叱精0.1g,加乙醇溶解、定容至 100ml)。分光光度计,色度测量系统(自研)。1.2 实验内容1.2.1乙氧基黄叱精指示剂溶液的吸收峰 将乙氧基黄叱精指示剂溶液滴入不同pH值的溶液中,在分光光度计测量其吸收峰,见表1、图2。http://ng1.17img.cn/bbsfiles/images/2017/10/2016091607491323_01_1722582_3.jpg图1. 乙氧基黄叱精指示剂在pH环境的吸收曲线 图2显示,乙氧基黄叱精在不同pH值的溶液中的最大吸收峰是不同的。当pH值增加时,吸收峰向短波长方向移动。在pH1、pH2、pH3、pH4、pH5、pH6时,最大吸收峰的波长依次是480nm、480 nm、475 nm、465 nm、450 nm、445 nm。其中在pH3以后至pH6期间变化幅度较大,说明其变色阈值在该范围内激烈变化。pH6以后,即使pH增加,最大吸收峰也是445nm,最大吸收峰保持不变,说明分子结构在pH6以后已经保持稳定,不在变化。表1. 不同PH环境下乙氧基黄叱精指示剂最大吸收峰的变化 PH 波长 1 2 3 4 5 6 7 8 9 10 11 12 445 1.12468 1.43217 1.40751 1.37924 1.23078 0.97832 1.32918 0.93405 1.19237 1.24271 0.94790 1.01212 450 1.21149 1.54126 1.49826 1.43608 1.23844 0.96863 1.31602 0.92405 1.17872 1.22770 0.93672 0.99734 455 1.28742 1.63532 1.57194 1.47602 1.22794 0.94405 1.28232 0.89929 1.14608
各位老师,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲胺磷、乙酰甲胺磷、氧乐果出的峰非常低,有时几乎看不见。更换了新称管那些还好一点,进样几次后又几乎不怎么出峰了,有什么办法解决下吗?
乙酰二茂铁的合成目的原理实验目的 1 通过乙酰二茂铁的制备,了解用Friendel-Crafts酰基化反应制备非苯芳酮的原理和方法。2 学习柱色谱分离提纯产品和薄层色谱跟踪反应进程的原理和操作方法。实验原理 二茂铁又名双环戊二烯基铁,是由2个环戊二烯负离子和一个二价铁离子键合而成。一般认为,以乙酸酐为酰化剂,三氟化硼,氢氟酸,磷酸为催化剂,主要生成一元取代物;如用无水三氯化铝为催化剂,酰氯或酸酐为酰化剂,当酰化剂与二茂铁的摩尔比为2∶1时,反应产物以1,1′-二元取代物为主。二茂铁及其衍生物的分离最好是用层析法。本实验用柱色谱分离提纯产品,可用薄层色谱法跟踪反应进程,柱色谱和薄层色谱均属于吸附色谱,柱色谱分离提纯是根据二茂铁,乙酰二茂铁和1,1′-二乙酰基二茂铁对活性氧化铝吸附能力的差异而进行分离提纯。用薄层色谱跟踪反应进程,根据二茂铁和乙酰二茂铁的斑点大小可以了解乙酰化反应的进程。仪器药品 5ml圆底烧瓶,克莱森接头,干燥管,电磁加热搅拌器,30cm色谱柱(自制),30×100mm载玻片,离心试管50ml烧杯,玻璃钉漏斗,吸滤瓶,锥形瓶,氮气袋,250ml烧杯二茂铁,乙酸酐,85%H3PO4,25%NaOH,二氯甲烷,棉花,洗净的砂,Ⅲ级活性氧化铝,己烷,醇,硅胶,0.5%羚甲基纤维素,干燥氮气。过程步骤 一、乙酰二茂铁的制备称取100mg(0.54mmol)二茂铁,放入5ml圆底烧瓶中,加入2.0ml醋酸酐。装上带有干燥管的克莱森接头。水浴温热并搅拌使二茂铁溶解。移去水浴,打开塞子迅速加入3ml 85% H3PO4,使反应液变成深红色,室温下搅拌1.5h,在反应期间定期用毛细管在液面上吸取2滴左右反应液放入具塞小试管中,假如10滴二氯甲烷,所得溶液用薄层色谱法展开,以了解反应进程。当二茂铁的斑点很浅时,表示反应基本完成。将反应液滴入盛有1g碎冰5ml烧杯中,滴加25%NaOH中和恰至碱性,得到大量桔黄色沉淀。充分冷却后抽滤,1ml冷水分几次洗涤沉淀,抽干,干燥后称重约110~120mg。二、乙酰基二茂铁的柱色谱法分离(1)干法装柱将粗产品溶于0.5ml二氯甲烷加入300mgⅢ级活性氧化铝,振荡均匀得浆状物。在通风橱中,在干燥氮气下除去溶剂至恒重,得到松散的颗粒状物,精确称取1/2用作柱色谱分离。将自制的1.5×30cm色谱柱洗净,干燥,柱底铺一层玻璃棉或脱脂棉,再铺一层约5~8mm厚的砂,填平。称取5gⅢ级活性的中性氧化铝(60~80目),通过漏斗将氧化铝装入柱管内,轻敲柱管,使之填均匀。将精确称得含有1/2产品重的氧化铝装入柱内,顶部盖一层约5mm厚的砂子,使氧化铝顶端和砂子上层保持水平。(2)洗脱用己烷作洗脱剂从柱顶加入,缓慢滴入己烷逐渐展开得到黄色、橙色分离的色谱带。黄色的二茂铁带首先从柱下流出,用己称重的锥形瓶收集洗脱溶液。当黄色谱带完全洗脱下来时,改用体积比为1∶1的二氯甲烷己烷混合物洗脱,同时橙色带往下移动,逐渐改变溶剂的比例到体积比9∶1二氯甲烷己烷混合溶剂时,则将橙色色谱带完全洗脱下来,用另一只已称重的锥形瓶收集洗脱液。最后改用体积比为9∶1二氯甲烷甲醇洗脱时,可以看到很淡的,很少量的,棕色色带向下移动,将该洗脱液另行收集。(3)收集产品在通风橱内,各组分洗脱液分别在水浴上蒸馏,回收溶剂。浓缩后的溶液放置冷却析出结晶,将产品放在盛有石蜡片的干燥器内至恒重。可回收到未反应的二茂铁20~22mg;得到乙酰二茂铁80~90mg 1,1′-二乙酰基二茂铁少于2mg。分别测定熔点。注意事项1.二茂铁需经升华或用石油醚(30~60℃)重结晶纯化。2.仪器应是充分干燥的。3.乙酸酐是临用前经重新蒸馏的。4.吸附剂的活性与其含水量的关,含水量越低,活性越高。氧化铝放入高温炉中(300~400℃)烘3h得无水物即Ⅰ级氧化铝。Ⅲ级氧化铝可用Ⅰ级活性氧化铝加入重量的6%的水而得到。如所用氧化铝活性过强会使产品不易洗脱,浪费较多的溶剂。5.这里是考虑到柱色谱的容器。一般粗产品重75mg以上都仅取1/2作柱色谱分离。6.二茂铁易升华,故测熔点时要封管。熔点的文献值:二茂铁为173℃,乙酰二茂铁为85℃,1,1ˊ-乙酰基二茂铁为130℃。分析思考1. 二茂铁乙酰化反应的机理怎样?2. 怎样利用薄层层析判断乙酰化反应的进程?3. 乙酰二茂铁在石油醚和乙醚中溶解度哪个更大?为什么?4. 柱层析分离二茂铁衍生物时,如何选择展开的溶剂? [img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52002_1632583_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52003_1632583_3.gif[/img]
化妆品中呋喃香豆素类(三甲沙林、8-甲氧基补骨脂素、5-甲氧基补骨脂素)和欧前胡内酯的检测方法
大家好,我现在在做乙酰乙酸甲酯气相色谱检测,用同一根色谱柱进样,有时候时两个峰,有时候是一个峰到底是什么原因?乙酰乙酸甲酯有互变异构体,在运行过程中有出现两个峰的可能。 现在能找到乙酰乙酸甲酯和乙酰乙酸乙酯的国标。乙酰乙酸甲酯的国标中是一各峰,而乙酰乙酸乙酯是两个峰,这两个东西的性质是相似的,为什么会出现不同的峰形呢?
各位兄弟姐妹:在下急需甲氧基溶纤剂乙酸酯,不知道在哪里能买到.
GBT 24800.3-2009 化妆品中螺内酯、过氧苯甲酰和维甲酸的测定 高效液相色谱法
我们公司做毒死蜱,合成工艺第一步是三氯乙酰氯和丙烯腈加成氯苯为溶剂,要根据标样的归一情况来确定投料量。标样里面丙烯腈大约10%三氯乙酰氯3.5%氯苯85%左右,之前一直是这个数据,今天突然三氯乙酰氯就变成2.8左右了。色谱条件是,进样口260,柱温90,检测器260。载气氮气,氢火焰检测器,柱子是SE—30非极性柱,用的空气发生器。仪器是国产的福利GC9790②。还有个问题就是氯苯有时候会出平顶峰,进样量0.2微升不到都平顶。以上两个问题还请各位大侠指导一下。

[color=#444444]用的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],乙醇和乙酰丙酸反应生成乙酰丙酸。乙醇和乙酰丙酸乙酯的峰分不开。柱前压0.15mpa.柱温200检测260注样260应该怎么调才能把峰分开?乙酰丙酸沸点245,乙酰丙酸乙酯沸点203[/color][color=#444444][img=,600,800]https://ng1.17img.cn/bbsfiles/images/2019/07/201907100951384017_3086_1843534_3.jpg!w600x800.jpg[/img][/color]
我第一次测土木香含量,哪位做过这个,土木香内酯和异土木香内酯的出峰时间是怎样的?色谱图是什么样的?请各位老师帮帮忙,谢谢
761做农残,之前做了大半年,柱子氧乐果不出峰了,没办法截了快两米,终于好了。可是发现做了两批样一百多个样,出峰面积又小了。走丙酮溶剂发现有小杂峰(面积很小,50左右。破罐子,破摔。各种折腾。用丙酮洗(进4微升),甲醇洗(进4微升)。老化的时候,进溶剂,甲醇,正己烷,丙酮轮着。结果更惨了,本来氧乐果1微升能出2000多的。现在就八百了。乙酰甲胺磷本来好好的,也少了很多。之前也发现,柱子老化后。本来不拖尾的峰,拖尾了。问各位老师,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱,走溶剂空白,能起到清洗作用吗?还是我用的溶剂体积太大了,能用甲醇吗?以后这种情况要每次做了,截了柱子后,再老化吗?
虽然不是特别的了解这个,但是有一次去饭馆吃饭,恰好遇见一个大厨在前台打电话要采购东西。我就顺便看了一眼他手中的纸条,发现上面写的是“内酯”不知道具体是什么,但凭直觉我觉得估计是添加剂吧现在饭馆里的菜的卖相,色泽什么的,包括香气,我觉得都很值得怀疑。能少去外面吃饭就少去吧,家常菜不家常啊也许某天这些东西也会被揭露吧,到时候恐怕又是食品安全的大问题。现炒现制的食品,现在还算是盲点吧不知道我的想法是对是错,请大家指正
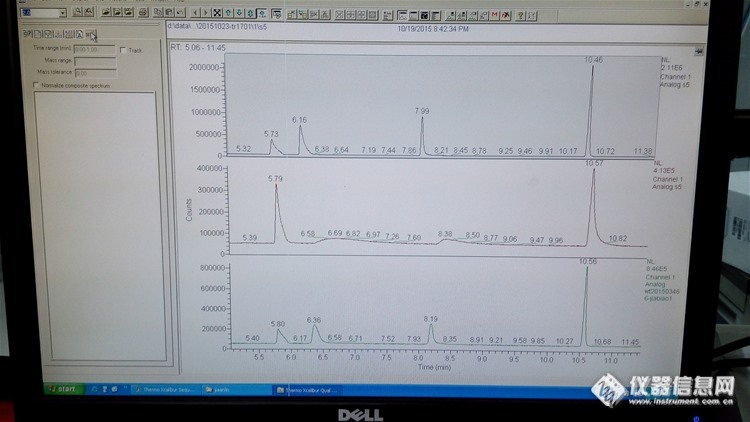
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
混酸法瓷质阳极氧化工艺的应用与改进 --------------------------------------------------------------------------------发布时间: 2007-12-11 10:56:25 浏览次数: 7 1 前言 瓷质阳极氧化是在铝或铝合金的表面,获得光滑的类似于搪瓷般的不透明膜的过程。膜的颜色一般为乳白色,在经过着色工序后可着上各种色泽。瓷质氧化膜具有良好的装饰性、耐蚀性和电绝缘性,因此,广泛应用于家用电器、仪器仪表和日用品[1]。但是,在工业生产中,仅能生成白色膜是不能满足实际需求的,后续的着色(不论是化学着色还是电解着色)必须有单独的工序、设备和操作人员,因而提高了生产成本,如何才能用一步法获得某种色泽的瓷质氧化膜是许多生产厂家都十分关注的。本文在查阅了近年来的有关文献,把氧化与着色相结合,在大量配方筛选的基础上开发出了一种一步法获得淡黄色瓷质氧化膜的工艺,省去了着色工序,明显地降低生产成本,获得了工业应用。2 瓷质阳极氧化工艺 2.1 工艺流程 铝材→化学除油→水洗→化学抛光→水洗→瓷质阳极氧化→水洗→封闭→晾干 2.2 化学除油 采用自制的化学除油溶液: NaOH 10~15g/L Na2CO3 30~50g/L Na3PO412H2O 25~40g/L 十二烷基硫酸钠 0.4~0.6g/L 除油温度应在60~80℃,除净以水洗后铝材表面无水成股流下,水膜均匀覆盖在表面为准,时间一般应在5~10s。 2.3 化学抛光 采用自制的化学抛光液,应注意化学抛光的温度和时间,防止过腐蚀。温度在85~90℃,时间约为10s。 2.4 瓷质阳极氧化 铬酐 30~35g/L硼酸 硼酸 6~10g/L 草酸 8~10g/L SnSO4 1.0~2.0g/L 添加剂 3~5g/L 温度 40~50℃ 电压 30~36V 电流密度 1.0~1.5A/dm2 时间 30~40min 其中:添加剂由几种多羟基酸和某种金属盐复配而成。 2.5 封闭 用沸水封闭约15min或重铬酸钾溶液封闭约10min。重铬酸钾还有增强黄色色泽和提高耐蚀性的作用。 2.6 工艺条件分析 2.6.1 电压和电流 电压和电流密度太小,着色性能差,甚至完全不能着色;电压和电流密度太大,膜层色调发暗,光泽度不好。因此,电压、电流密度应控制在工艺规范中,最佳电压在35V,电流密度1A/dm2。 2.6.2 SnSO4的用量 SnSO4的用量是本工艺中的关键条件,应控制SnSO4的含量在1.0g/L左右,如要求色泽较深,可适当增加用量。 2.6.3 添加剂的用量 添加剂由几种多羟基羧酸和某种金属盐复配而成,能明显提高氧化膜的瓷质感,改善氧化膜的色泽,同时提高着色速率。添加剂的用量过低,氧化膜发暗,光泽不好;然而,添加剂的用量超过5g/L,光泽度的提高效果已不显著,而成本却上升了。因此,添加剂用量应控制在3~5g/L为好。 3 结论 (1)本瓷质氧化工艺通过SnSO4和添加剂的加入,一步法获得了淡黄色的瓷质氧化膜层,降低了生产成本。 (2)本工艺不需机械抛光,只进行化学抛光即可达到光泽度的要求。 (3)本工艺氧化膜的各项指标,如外观、耐蚀性能、耐磨性能等均可达到国家标准,因此适用于家用电器、仪器仪表和日用品的表面装饰。资讯来源: 混酸法瓷质阳极氧化工艺的应用与改进 发布人: 全球电镀网
上来几批样品,标品中甲胺磷,乙酰甲胺磷,氧乐果就消失不见了,每次维护完仪器(更换衬管,清洗离子源,老化色谱柱)才能恢复,前面以为是仪器有污染了吸附,这一次都还是,就5-60个样品,后面就自动消失了。用的安捷伦7000
GC450瓦里安气相,PFPD检测器,DB1701(30*0.25*0.25)柱,进样口温度250℃,检测器300℃;程序升温:80℃保持1min,20℃/min升至130℃,5℃/min升至200℃,15℃/min升至250℃,保持1min,共21.83min。乙酰甲胺磷标液是新打开的,浓度100ug/mL,用丙酮配制成1.0ug/ml进样,乙酰甲胺磷不出峰,但配制的二唪农0.1ug/mL出尖峰。谁做过乙酰甲胺磷,用什么溶剂,它在0.1ug/mL时就出峰?
岛津的2014C[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],FPD检测器,1701柱子,高惰性衬管,之前做有机磷混标的时候用丙酮溶剂0.1ug/ml敌敌畏,甲胺磷,乙酰甲胺磷,氧乐果,都出峰,除了乙酰甲胺磷出峰峰型都还好,可是这次做,这四种就是不出峰了,衬管也换了,柱子也裁了,还是不出峰,又新开标液重配还是不出峰,然后开始大浓度5ug/ml,单标进样,都出峰了,但是敌敌畏和氧乐果的峰面积偏小,直至低浓度的敌敌畏和氧乐果的1ug/ml的出峰,然后0.5ug/ml,就不出峰了。然后用黄瓜基质配做溶剂配的混标,敌敌畏0.2,甲胺磷和乙酰甲胺磷、氧乐果0.1ug/ml竟然都出峰了,而且除了敌敌畏峰小点,其他出峰都还可以,连续进了四五针峰面积也差不多,同时用丙酮配四种一样浓度的混标还是不出峰,这是啥原因造成的的?(PS:新换衬管我连续进了七八针大浓度的,然后再进的小浓度的都不出峰)
RT,求购9-五代甲氧基酰氯、fluorenylmethoxycarbonyl联系方式QQ:15486575e-mail:rjl2000cn@yahoo.com.cn
[color=#444444]用六氯环三磷腈和乙醇钠合成六乙氧基环三磷腈,重结晶后测其红外,发现在2660处出现一个峰,查了很多资料,也觉得这个产物在此处不该出峰啊 ,请大神来解析解析这个六乙氧基环三磷腈的红外图[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2019/0325/bw196h8429997_1553514396_147.png#opennewwindow[/img][/color]
在做蔬菜农残时,以前有机磷混标(1mg/L)的各峰高均差不多高。现就不一样了,乙酰甲胺磷的峰几乎是原来峰的10%了,而甲拌磷比原来的倒高了,为什么?真的纳闷呀
AAbrine 相思豆碱O-Acetyl-3,6-di-O-β-D-xylopy-rano- astragaloside O-乙烯 3,6-双氧-β-D- 吡喃木糖基绵毛黄芪甙Acetylastragaloside 乙酰黄芪甙N-Acetyl-D-Glucosamine N-乙酰氨基葡萄糖糖6''-acetylhyperoside 6''-乙酰氧基金丝桃甙Acetylshikonin 乙酰紫草素14-Acetyltalatisamine Achyranthan 牛膝多糖Acobreting D 短距乌头碱丁Acobreting E 短距乌头碱戊Aconine 乌头原碱Aconitine 乌头碱Aconosine 爱康诺辛Actein 黄肉楠碱Actinodephnine Acuminatin Acuminatoside Adenanthin 腺华素Adenine 腺嘌呤Adenoside 腺苷Adynerin 欧夹竹桃甙乙Aescin 七叶皂甙Aesculetin 七叶素(七叶内脂,秦皮乙素)马栗树皮素Aesculin 七叶甙 马栗树皮甙Agaricic acid 落叶松覃酸Agaricus blazei murrill P.E 姬松茸提取物Agrimophol 仙鹤草酚Ajmalicine(δ-Yohimbine) 阿吗碱,δ-育亨宾碱,阿吗里新Ajmaline 阿马林Akebia saponin D 木通皂甙 DAlantolactone 土木香内酯Albopilosin A Aleuritic acid 苏式-紫胶桐酸Alfalfa P.E 紫苜蓿提取物Alizarin 茜素Alkaloids 罗勒生物碱Allantoin 尿囊素Allasecurinine 别一叶秋碱Allantolin Allicin 大蒜素α-Allocryptopine α-别隐品碱Alloisoimperatorin 别异欧前胡素Alloxanthoxyletin Allose 阿罗糖Aloeemodin 芦荟大黄素Aloe-saponol Aloin 芦荟甙Aloesin 芦荟苦素Aloperin 苦豆碱Alpinetin 山姜素Amentoflavone 1- Amino-cyclopropane-1-acid hydrochloride 1-氨基环丙烷-1-羧酸盐酸盐Amethystoidin A 香茶菜甲素Ampelopstin 福建茶素Amygdalase 苦杏仁酶Amygdalin 苦杏仁甙Aamylase 淀粉酶β-Amyrin β-香树脂醇α-Amyrin acetate α-香树脂醇乙酸乙酯β-Amyrin acetate β-香树脂醇乙酸乙酯β-Amyrin palmitate 棕榈酰β-香树酯Anabasine 毒藜碱Anagyrine 臭豆碱,安那吉碱Andrographis Paniculata P.E 穿心莲提取物Andrographolide 穿心莲内酯 穿心莲乙素Anemonin 白头翁素Angelica Dongquai P.E 当归提取物Anisodamine 山莨菪碱Anisodine 樟柳碱Anthocyanosides 花色素Anthraglycoside A 大黄素-6-甲醚-8-D-葡萄糖甙Anthraglycoside B 大黄素-8-D-葡萄糖甙Apigenin 芹菜甙元 芹菜素Apigenin-7-glucoside 芹菜甙元-7- 葡萄糖甙Apigenin-7-O-glucoside 芹菜甙元-7- O-葡萄糖甙Apigenin-8-O-glucoside 芹菜甙元-8- O-葡萄糖甙Apigenin-7-O-α-L-rhamnoside 芹黄素-7-O-α-L-鼠李糖甙Apiin 芹菜甙Apple Extract 苹果提取物Araloside A 楤木皂甙 AArbutin 熊果甙Arctigenin 牛蒡甙元Arctiin 牛蒡子甙Arctinal 牛蒡子醛Arctinol b 牛蒡子醇-bAristolactone 马兜铃内酯Aristolochic acid A, D 马兜铃酸A,DAristolochic acid B, C 马兜铃酸B,CArmillarisin A 亮菌甲素,假蜜环菌甲素Arteannuic acid 青蒿酸Arteannuin 青蒿素Artemisinin 青蒿提取物α-Artemether α-蒿甲醚β-Artemether β-蒿甲醚Artemetin 青蒿亭Artemisinic acid 春活力 青蒿酸Arundoin 芦竹素Asarinin 细辛脂素α-Asaron α-细辛醚β-Asarone β-细辛醚Asiatic acid 积雪草酸Asiaticoside 积雪草甙Astilbin 落新妇甙Astragaloside 黄芪甙Astragalin Astragaloside II 黄芪皂甙 IIAstragaloside IV 黄芪甲甙Astragalus Root P.E 黄芪提取物Atisine chloride Atractydin 苍术素Atractylon 苍术酮Atraotydin 苍术甙Atropine 阿托品Aucubin 桃叶珊瑚甙Avacularin 扁蓄甙
乙酰乙酸甲酯项目,HG/T 4479-2012标准里只有一个乙酰乙酸甲酯峰,但是用安捷伦HP-1分离时出现2个峰,怎么调整参数可以只出现1个峰?







 我要推广仪器
我要推广仪器
 下载APP
下载APP