用的是浙江温岭9790的气象色谱仪,毛细管柱子。同一样品甲基三甲氧基硅烷(其沸点102~103),监测器进样器温度150,柱子温度分别为60、80、100、120度,恒温打样,发现低温主峰后小峰成M型且峰高降低(为什么???),高温主峰后小峰分离紧凑,100度分离最好。请问专家,沸点和柱温何关系才能达到最佳分离效果?低温为什么出现M峰???
【序号】:2【作者】: 郭乃妮郑敏燕杨连利【题名】:超声条件下季铵盐3-十二烷氧基-2-十八酰氧基丙基三甲基氯化铵的合成研究【期刊】:皮革与化工. 【年、卷、期、起止页码】:2017,34(06)【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2018&filename=PGHG201706003&uniplatform=NZKPT&v=FWkDKY1K2RpbTCMi7HzpBtcfxqQrFRqm95YR3lJywpqICHhucEksIDY_X_QTFQWc
购买丙基三甲氧基硅烷等类的试剂,怎样才能方便快速买到?谢谢
这几天一直围绕着三甲基氯硅烷,这东西太不稳定,又是手动进样,分流又不可控,纯度从80%到95%不等,不知道什么原因。另,是不是三甲基氯硅烷与乙醇也反应啊,或是乙醇中有水,我将三甲基氯硅烷与色谱纯的无水乙醇以1:1混合后,放置4小时,溶液呈绿色,而三甲基氯硅烷呈浅红色。不知道什么原因!是不是三甲基氯硅烷反应生成的盐酸与进样针的铁反应了?因为三价铁是棕色的,二价铁是绿色?推测请神人帮忙,谢谢!
有关单位: 经国家食品药品监督管理局化妆品审评专家委员会审核,拟批准“二甲氧基甲苯基-4-丙基间苯二酚”和“聚甲基丙烯酰基赖氨酸”作为化妆品原料使用。现公开征求意见,请于2011年6月27日前将反馈意见电子版发送至chenzh@sfda.gov.cn。 附件:1.“二甲氧基甲苯基-4-丙基间苯二酚”技术要求 2.“聚甲基丙烯酰基赖氨酸”技术要求 国家食品药品监督管理局食品许可司 二〇一一年六月十五日
没有经验,虚心求教!最近用高回收率Agilent样品瓶进行生物样本的三甲基硅烷化衍生,最后加入正己烷离心后直接进样,总体积400微升,发现污染严重,导致批量样品无法检测。大家在最后环节都是怎么处理的?转移到微量内插管里?还是仍然留在衍生瓶里进样?我试着转到内插管里,但是谱图有较明显差异,难道用去活玻璃内插管?
求购咨询苯基三甲氧基硅烷与辛基三乙氧基硅烷的色度检测标准和仪器,或者告诉下哪里有权威的检测机构也好的。谢谢!
我用气相色谱分析脂肪醇,用吡啶溶解,再加三甲基硅烷时,出沉淀,不分层!继续加六甲基二硅胺烷时,出现分层,下面是沉淀,请问沉淀是什么物质?请问有知道的吗?急!
[color=#444444]测定白油中环己基甲基二甲氧基硅烷的浓度,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]外标法,重复性很差,想请教一下问题在哪里?[/color][color=#444444]色谱条件简单如下:岛津GC-2014,无自动进样器[/color][color=#444444]柱温:100℃;进样口200℃;检测器300℃;[/color][color=#444444]程序升温:100℃(1min)--5℃/min升至150℃(0min)—25℃/min升至200℃(5min)[/color][color=#444444]色谱柱为非极性毛细管柱[/color][color=#444444]分流比:80:1[/color][color=#444444]样品采用正己烷稀释10倍后分析,质量浓度约15%,色谱分析重复性很差,做过此类分析的高手给些建议,谢谢![/color]
我用的是10%OV-101填充柱,检测三甲氧基硅烷和四甲氧基硅烷,出的峰较宽,且四甲氧基硅烷经常好出拖尾峰,是不是和柱效有关?柱子老化后出和峰还是较宽。
聚丙烯酰胺和十六烷基三甲基氯化铵分析检测,用什么仪器?
有哪位大侠做过三(三甲基硅烷)硼酸酯或三(三甲基硅烷)磷酸酯的分析啊?用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的话选用何种柱子合适啊?
用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
关于有机硅分析微量水分,甲苯,甲基三乙氧基硅烷,氯化苯混合溶液怎么配[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]?
求 JIS K9555-1992 三甲基氯硅烷标准
三甲基硅烷醚的结构式是什么?有没异构体?别称?CAS号?
[font=Times New Roman]三甲基硅烷化方法是糖的羟基衍生化的主要方式之一。优点是:衍生物挥发性强,制备快速、简便。衍生化反应可在室温下几分钟完成,它可适用于醛糖、酮糖、甙、糖醇、糖醛酸和脱氧糖等的衍生化。缺点是:由于各种单糖异构体和不同大小环的特殊单糖的存在,造成色谱峰多于组分单糖的数目,从而导致混合物的定性定量分析复杂化。[/font]那么如何用内标法/外标法对同种糖的不同峰定量呢 以求出该糖总浓度?
[em09506]三甲基氯硅烷的检测方法?在网上查到说是用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测,但没有具体说明的(比如载气是什么?等),希望有知道的朋友帮帮忙吧~谢谢了!
新手小白,请问哪位老师做过GBT34263-2017工业用羟丙基甲基纤维素 中甲氧基、羟丙氧基含量的测定? 仪器方面该如何配置?顶空进样器?液体进样器?为什么使用顶空瓶做前处理?
请问各位大侠谁知道三甲基硅烷基键合硅胶的液相色谱柱,请告知是那家公司的产品或经销商,多少钱? 要进口的,请经销商帮帮忙,多谢了。
3-氨丙基三乙氧基硅烷,分析纯就可以了,谢谢。

测N-(甲氧甲基)-N-(三甲基硅甲基)苄胺我采用岛津[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],柱子为wax(聚乙二醇),所设参数SPL 290度,DFID 290度,柱温40度下保留1 min再以10度每分升温到240度,直接进样走到中间在某一峰后出现基线严重漂移,但基线可以回到零点,用甲醇稀释后基线漂移没有那么明显,而此处也显示有另外一个物质存在,想请教高手出现这种状况是什么原因?是物质沸点太高参数选择不对还是物质与柱子极性不匹配?N-(甲氧甲基)-N-(三甲基硅甲基)苄胺的沸点为76 º C (0.3 MMHG)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181274]N-(甲氧甲基)-N-(三甲基硅甲基)苄胺色谱图(甲醇溶解).doc[/url][img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911051117_181361_1618994_3.jpg[/img]
三甲基氧化铵的测定方法,非常急,请多帮助!
群友问:用GB 5413.25-2010 第二法气相色谱法做肌醇,实验室买的三甲基氯硅烷和六甲基二硅胺烷有本底干扰,不知道老师们这两个试剂买的是什么牌子的?群友一:国药的群友二:三甲基氯硅烷,扫尾剂级别,100ml,国药有的,六甲基二硅胺烷,国扫尾剂级别,25ml,也是国药的;这两个试剂SIGMA有专用的衍生剂级别的。非常感谢乳制品检测之家群友岑老师、人气老师的分享!
请问大家有没有检测过三甲基胂氧(TMAO),这个物质需要进口,询价很贵,而且货期很长,大家有没有谁有买过这个物质?
今天配制过程如下: 移取100uL三甲基氯矽烷(TMS)入1.8mL进样小瓶,然后再移取300uL六甲基二矽烷(HMDS)入之,一切正常,但是再移取900uL吡啶入之时,先出现“白雾”,然后再出现固态“沉淀”。 奇怪的是这沉淀漂在上面,振摇,沉淀减少,而且底部的衍生试剂开始澄清。 这正常吗?
填充柱填充的是10%OV-101,分析三甲氧基硅烷时,出的峰的顶部是歪峰,是不是和固定液涂布的多少有关系?
由于手上没有2,3,4-三甲氧基乙酰苯(2,3,4,-trimethoxyacetophenone,C11H14O4)的对照品,所以想求助大家有没有它的光谱图,有的请发一份上来好吗?谢谢大家帮忙啦,
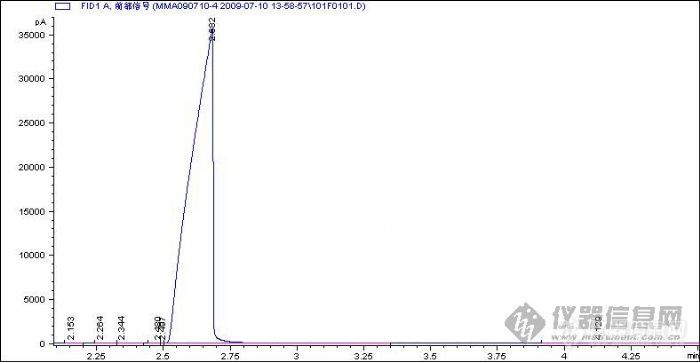
各位,请帮助参考下,看看在现有的条件下,有什么办法能够提高各组分的分离度?进样物质:甲基丙烯酸甲酯(AR级)溶剂,可能含有的组分有:甲醇(),丙酮,丙烯酸甲酯,异丁酸甲酯, 甲基丙烯酸乙酯,а-羟基异丁酸甲酯.仪器:AGILENT7890A ,FID, 毛细管DB-WAX: 30m*320mm*0.25mm,ASL进样:1uL.操作条件:分流比:50:1.压力:7psi,27.506sec/min.初温35度,保持0.1分钟,然后以4度/分钟升至50度,再以15度/分钟升至150度,保持10分钟.色谱图:图1:放大图[img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907101554_159431_1625521_3.jpg[/img]图2:缩小图1[img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907101554_159432_1625521_3.jpg[/img]图2:缩小图2[img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907101555_159433_1625521_3.jpg[/img]问题:1在图1中,进入的是溶剂甲基丙烯酸甲酯,为什么会出现主成分的色谱图是前伸峰,而有时候,我在做其它的溶剂的时候,会出现后延峰?2在图2中,除上述的分离方法外,在没有办法改变硬件的情况下,是否有别的办法提高分离度.应该还需要变化那些条件,如何去变化?3在图3中,组分(未知)4.129和9.997是否拖尾太历害,有什么办法改进?(确认不是鬼峰)
我用的是10%OV-101的填充柱,分析三甲氧基硅烷时峰的顶端不对称,右边的峰向下坡的很一些。请问是什么原因?







 我要推广仪器
我要推广仪器
 下载APP
下载APP