有人做过辛菌胺吗?都是用什么仪器做的?
今天我看到这样一个新闻信息与大家分享一下:亲贝网讯 5月31日消息,四川省保护消费者权益委员会(下称“四川消委会”)5月30日发布《关于婴儿配方奶粉比较试验的报告》。报告显示,标称明一(福建)婴幼儿营养品有限公司生产的一款明一牌金智婴婴儿配方奶粉检出磺胺多辛。农业部《动物性食品中兽药最高残留限量》(2002年235号公告)中的有关规定。只对磺胺二甲基嘧啶(25ppb)和磺胺总量(100ppb)进行了规定。并未对磺胺多辛进行明确的进行规定。再次网上有关的资料中是这样说的:【相关资料显示,小于2个月婴儿禁用磺胺多辛。“由于磺胺药可与胆红素竞争在血浆蛋白上的结合部位,而新生儿的乙酰转移酶系统未发育完善,磺胺游离血浓度增高,以致增加了核黄疸发生的危险性,因此该类药物在新生儿及2个月以下婴儿的应用属禁忌。”】这里所说的禁忌,是什么意思?为什么到媒体的口中就成了禁用了啊?”被出15ppb,牛奶一般做成奶粉需要浓缩8倍左右,也就是说在收奶的过程中需要检测到1-2ppb的方法才可能是15个ppb,对于牛奶中检测磺胺多辛要检测到1-2ppb的检测能力,我想做检测的大家都应该知道有多难。只能上精密仪器,但是仪器有检测时间都比较长,需要复杂的前处理工作。而牛奶要放在那一二天就不知道能不能喝了。”最后发表一点我个人的观点,牛奶出在牛身上,牛在奶农手中,下药的都是兽医。标准中国家定的,牛奶是厂里生产的,喝奶的都是我们。不合适是媒体曝光的,检测是政府做的。达到标准了,被曝光了,奶不能喝了,牛不能养了。你查磺胺,我用喹诺酮;你查喹诺酮,我用庆大;你查庆大,我用林可;你查~~~~,我用~~~~~,永远都是猫捉老鼠,很好完,没有头。最后苦的是企业(不知道怎么查才安心),是喝奶的人啊(不知道喝谁的奶安全)。
[color=#444444]三辛胺、三辛胺的盐酸盐与正辛醇组成的混合液,能用液相色谱分析吗?若不能用上述方法分析,那应该用什么方法,求各位大神指教,谢谢[/color]
请问葡辛胺可以用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测吗?请教一下具体检测方法
[color=#444444]目前在用辛胺和乙腈做流动相走梯度洗脱,来分析样品,主要是想做[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]。在用流动相分析样样品时主要遇到下面问题:[/color][color=#444444]1、基线不稳,噪音较大;[/color][color=#444444]2、走空白和样品时,都会出现一个大包包,然后,后面有几个峰依次出来,[/color][color=#444444]这样由于这个包包的问题就没法上质谱,后面的杂质检测不出来,没法定性。请教一下各位有什么办法没啊[/color]
[color=#444444]现在在做原料药的残留溶剂方法开发,十几种溶剂中的三乙胺、异辛烷、苯三个溶剂不管是调流速,还是调节升温速率,总是分不开,有经验的朋友帮帮忙出出主意,应该怎么优化啊[/color][color=#444444]现在用的柱子是中等极性的Agilent DB-624 30m*0.530mm*3.0μm流速3.0ml/min,初始40℃,15min,一点要分开的趋势都没有。[/color]
请教双胍辛胺醋酸盐的检测方法,液相或[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的方法均可
NY761-2008前处理,FPD检测器,DB1701柱子。乙酰甲胺磷、甲胺磷的响应值小,峰形难看,回收率也低,敢问高手,这两种农药的回收率最高可做到多少?什么条件下做得?最好有图谱,交流学习。
什么条件能出峰?用的~5的柱子乙酰甲胺磷看不到明显的峰,只有一个山丘似的不成峰,敢问做过的指点一下方法
检测一线的网友们,你们除了三氯氰胺,有测三聚氰酸二酰胺、三聚氰酸一酰胺、三聚氰酸吗?三氯氰胺的其他类似物?三聚氰酸有剧毒,并有刺激性。溶于热水。有人知道奶粉中加入的三氯氰胺纯度情况?有那些杂质?加入这种非法添加剂的稳定性情况?可能生成哪些新成分?
用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做敌百虫、辛硫磷、乙酰甲胺磷的效果不是很好,想尝试用用液相方法做,但不知道如何下手,请各位高人帮忙指点一下,万分感谢了!
用NY761方法做乙酰甲胺磷与甲胺磷的回收率,标液用丙酮配制,衬管用高惰性衬管,不是新的,是用过的,添加量为0.16ug/mL,甲胺磷实际测得为0.20ug/mL,乙酰甲胺磷为0.35ug/mL,回收率为125-216%,为何回收率这么高,是因为高惰性衬管受到污染了吗?还是必须用基质配标液。
氰化物检测的时候都是蒸馏完了先加入氯胺T,在加入异烟酸啉酮或者巴比妥酸,氯胺T都是要临用时现配,很麻烦,我听有人说可以不用氯胺T,可是反应的时候是和氯胺T反应生成氯化氰,那么就不能去除掉加氯胺T这一步了吗?有谁有更好的测定方法,或者更简单的比色方法呢?最好是用耐保存的试剂。想要一个生活饮用水新的检测方法,刚才搜索了,没搜到
在线GPC-GC-MS仪器,换的新柱测定有机磷,第一针甲胺磷敌敌畏均出峰,后面则进针均是甲胺磷不出峰,而敌敌畏出峰的原因
我想知道如何检测三辛胺和煤油混合物中各自的含量?
蔬菜中有机磷农药的检测,新换的衬管分别用用甲胺磷和乙酰甲胺磷单标饱和,然后接着做混标,发现甲胺磷和乙酰甲胺磷的单标和混标中的出峰时间不一致,相差挺大的,单标是3个月前配的(冷藏),以前也没出现过这种情况,请教各问大侠了,谢谢!
请问专家,羟胺和盐酸羟胺有什么区别,若有区别,有什么办法可使盐酸羟胺变为羟胺,请各位不吝赐教,衷心感谢!!!
我最近在用纯水系统里面添加了,五水硫酸铜和辛胺,水系:甲醇(95:5)紫外检测器215nm有时候做样会出现主峰保留时间提前一分钟。希望大家能够帮忙分析。急!急!急!急!急!急!急!急!急!急!急!急!急!急!急!急!急!急!
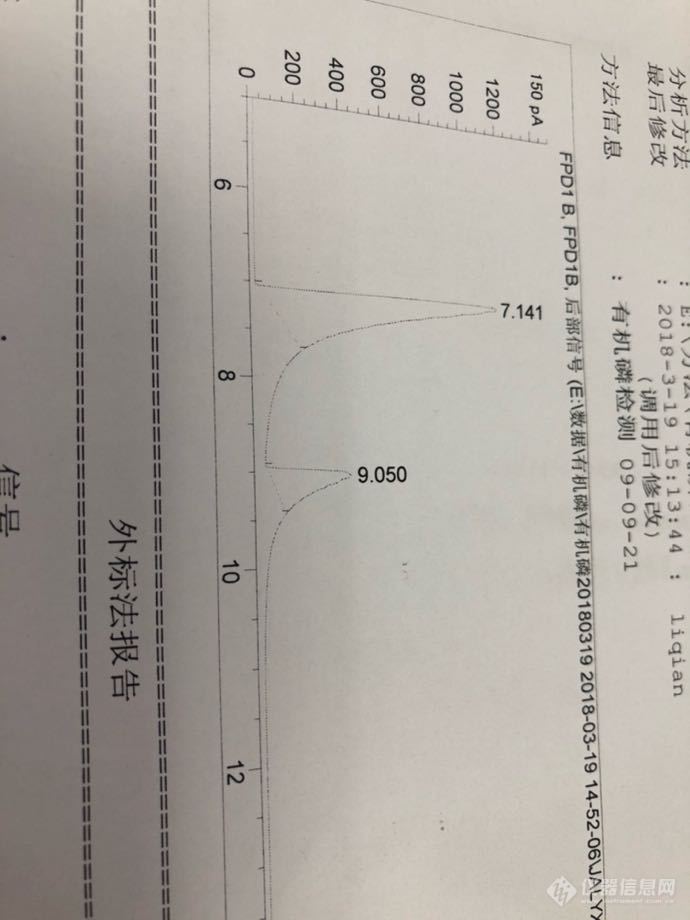
甲胺磷 乙酰甲胺磷拖尾严重…请问有啥解决办法吗?衬管也是新换的,柱头也割过一截了。方法也是之前用的。标准品也是新开的…请问有啥解决办法吗?[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2018/03/201803191602167875_8255_3379628_3.png[/img]
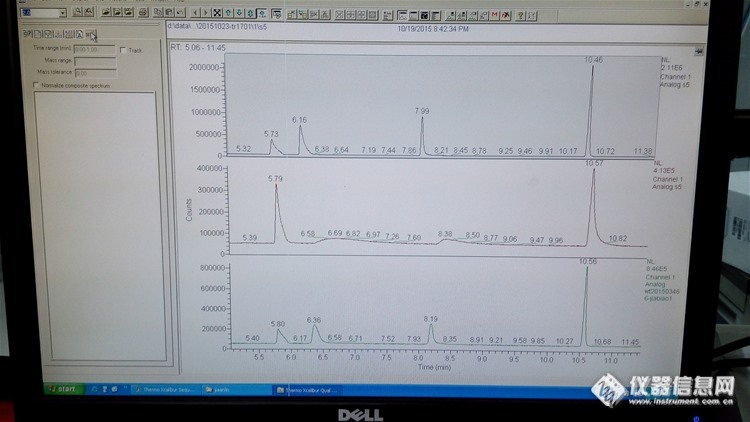
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
蔬菜中有机磷农药的检测,新换的衬管分别用用甲胺磷和乙酰甲胺磷单标饱和,然后接着做混标,发现甲胺磷和乙酰甲胺磷的单标和混标中的出峰时间不一致,相差挺大的,单标是3个月前配的(冷藏),以前也没出现过这种情况,请教各问大侠了,谢谢!
有哪位大神做过职业卫生的乙胺 乙二胺和环己胺的测定,我始终找不到乙二胺的峰,如果有做出来的,能发一下条件吗
最近做农残,发现0.5的农残混标中甲胺磷乙酰甲胺磷氧乐果峰小的可怜,我也按照论坛上的方法试了,截取了20厘米的柱子,换了新衬管和进样垫,重新配了1ug/ml的标样,还是那样,这几种物质峰特别小,请大家帮忙分析下原因,急!!!
各位老师,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲胺磷、乙酰甲胺磷、氧乐果出的峰非常低,有时几乎看不见。更换了新称管那些还好一点,进样几次后又几乎不怎么出峰了,有什么办法解决下吗?
最近在做水产品中磺胺类药物残留检测,我们用的标准是农业部958号公告-12-2007,主要做磺胺嘧啶,磺胺噻唑,磺胺甲基嘧啶,磺胺二甲基嘧啶,磺胺甲基异恶唑,磺胺多辛,磺胺异恶唑,磺胺喹恶啉这八种。发现这八种性质还是差别挺大的,其中磺胺嘧啶,磺胺噻唑还有磺胺喹恶林的回收一直做不好,估计也就30-40%,中间那几种回收倒是可以,不知道大家做的怎么样,希望能多多大家的宝贵意见,谢谢啦!

10,抽取5个版友);中奖名单:zengzhengce163(注册ID:zengzhengce163)馨语(注册ID:huangdm)mengzhaocheng(注册ID:mengzhaocheng)大川之子,纵横四海(注册ID:chuangu120)zgx3025(注册ID:v2844608)http://ng1.17img.cn/bbsfiles/images/2016/06/201606081538_596390_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/06/201606081538_596391_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================胺、苯胺和酚类物质方法:GC基质:标准溶液应用编号:101127化合物:二乙胺; 吡啶;吗啉;酚;苯胺;2- 氯酚; 二亚乙基三胺; 辛烷基胺; 1- 甲基-2- 吡咯烷酮; 2- 硝基苯酚; 2,6- 二甲基苯胺; 尼古丁; 2- 硝基苯胺固定相:DM-5 Amine色谱柱/前处理小柱:DM-5 Amine 30m x 0.32mm x 1.0u色谱条件:柱温:120 oC - 220 ℃, 10 ℃/min 载气:H2, 38 cm/sec, 120 ℃ 进样方式:分流, 25:1, 305 ℃ 样品:胺和苯酚类物质的水溶液, 1.0 μL, 22 ng 检测:FID, 6.4 x 10-11 AFS, 305 ℃文章出处:CCR00301关键字:胺,苯胺,酚类化合物,GC,DM-5 Amine,化工谱图:http://www.dikma.com.cn/Public/Uploads/images/CCR00301.png图例:1. 二乙胺;2. 吡啶;3. 吗啉;4. 酚;5. 苯胺;6. 2- 氯酚;7. 二亚乙基三胺;8. 辛烷基胺;9. 1- 甲基-2- 吡咯烷酮;10. 2- 硝基苯酚;11. 2,6- 二甲基苯胺;12. 尼古丁;13. 2- 硝基苯胺
有的水相加甲酸胺,有的加乙酸胺,有什么区别,什么时候加甲酸胺,什么时候加乙酸胺?如果正离子模式多,应该加哪个?
[font=微软雅黑]点击链接查看更多:[url]https://www.woyaoce.cn/service/info-4155.html[/url]乙草胺(acetochlor),即2-乙基-6甲基--N-乙氧基甲基-α-氯代乙酰替苯胺,是一种广泛应用的除草剂,人体暴露在乙草胺每日摄取容许量以上的环境下会对造成一定的潜在危害影响,并且目前还不能排除基因毒性的存在。[/font][font=微软雅黑]乙草胺因其毒性,被美国环境保护局定为B-2类致癌物,规定在1个月的监测期,在20个监测井所谓地下水中残留浓度不得超过0.1μg/L,kolpin等报道了美国在乙草胺的施用期的样品检测结果,1994年雨水和喝水中最大检出浓度分别是2.5、1.2μg/L。乙草胺作为玉米、大豆、棉花和果树的除草剂,在我国的年使用量已超多10000t(原药)。[/font][font=微软雅黑]丁草胺(Butachlor),2-氯-N-(2,6-二乙基苯基)-N-(丁氧甲基)乙酰胺,是选择性芽前除草剂,植物吸收丁草胺后,在体内抑制和破坏蛋白酶,影响蛋白质的形成,抑制杂草幼芽和幼根正常生长发育,从而使杂草死亡。在粘壤土及有机质含量较高的土壤上使用,药剂可被土壤胶体吸收,不易被淋溶,特效期可达1-2个月。[/font][font=微软雅黑]丁草胺具有挥发性,它对人体皮肤和眼睛有轻微的刺激作用,中毒的主要表现为消化系统与神经系统症状。轻者可引起胃肠功能紊乱,出现恶心、呕吐、吞咽困难、头晕、头痛等;严重者可引起麻醉作用,表现为头痛、头晕、无力、面潮红,酒醉状态,恶心、呕吐、呼吸困难、眼和呼吸道刺激症状和四肢麻木等。严重时可出现意识蒙眬、抽搐、昏迷、心室纤颤,呼吸停止而即刻死亡。[/font][font=微软雅黑]鉴科检测参考《DB21T 1546-2007 土壤中乙草胺和丁草胺残留量的测定 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法》,建立了利用全自动固相萃取仪(Fotector Plus)结合[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检测沉积物中乙草胺和丁草胺残留量的方法。在40mL丙酮-正己烷(4+1)提取后过滤,再用30mL丙酮-正己烷(4+1)复提2次,合并提取液。使用Auto EVA-08IR浓缩至2mL后 Fotector Plus全自动固相萃取仪净化,自动完成 SPE 柱活化、样品上样、淋洗、收集等步骤,收集液再氮吹浓缩、溶剂转换、定容后,用GC检测。[/font]
[color=#444444]三乙胺-丙酮-盐的混合液,三乙胺的含量在5%以下,请问有什么好的方法可以测定三乙胺的含量,在实验室用氢火焰毛细柱打可以出峰,但是如果含量特别低时担心[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]打不出,导致微量三乙胺检测不出来。查文献有溴酚蓝分光光度法、红外色谱,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]、电位返滴定法,以及混合液放入盐酸,用过量NaOH滴定未反应酸,溴酚蓝做指示剂等方法。请问这几种方法哪个比较准确,并且好操作,谢谢各位了![/color]
问题描述: 四烷基季铵盐(如四丁基硫酸氢铵、四丁基溴化铵、四丁基氢氧化铵等)在水中电离后,也形成了类似N+H(CH2CH3)3的结构N+(CH2CH2CH2CH3)4 ,这种结构也能有效的与Si-O-产生较强的静电作用,此类离子对用的比较少,但它的作用仅是掩藏Si-O-吗,它对物质的保留性能有何影响,实验中发现检测胺化物时,流动相中加入磷酸缓冲盐有增加物质保留的趋势,而加入三乙胺则会降低物质的保留能力? 解答: 四丁基氢氧化铵等离子对试剂确实不如辛磺酸钠等极性基阴离子的离子对试剂用得普遍,原因是酸类极性物质很容易通过降低pH值的方法提高在反相色谱中的保留能力,降低pH可抑制酸的电离,使酸处于中性状态而与疏水碳链的作用力增强。而碱类分析物则受硅胶基质pH上限的严重影响(以前上限是8,后来抬到10,直到最近杂化硅胶才将pH上限提到12左右)。 铵盐类离子对试剂确实有辛磺酸钠等所不具有的屏蔽硅醇基作用,但其主要作用还是疏水端和疏水固定相结合,外露的阳离子亲水端和酸阴离子作用,从而提高其保留能力。当然同样还有第二种解释机理,离子对试剂先和分析物结合,掩藏极性基,从而提高极性分析物在反相色谱中的保留能力。而且丁基比辛基疏水性差,这种情况下,认为后面的结合机理占上风的可能性更大。 检测胺类化合物,加入三乙胺预先和硅醇基结合,胺和硅醇基作用被三乙胺取代,保留下降是肯定的。







 我要推广仪器
我要推广仪器
 下载APP
下载APP