求购四甲基四氢环四硅氧烷,工业级别的即可,急!!!电子信箱:zongbin1783@yahoo.com.cn
最近公司突然需要分析桥式四氢双环戊二烯的纯度,不知道谁有这方面的资料,帮帮忙分享下。
[color=#444444]求问ESI-ms是否可能把 苄基作为保护基的糖的改造产物打成碎片~[/color][color=#444444]今天所得到的质谱峰非常明显(1163和455,相对丰度是100和15),但不是目标产物的分子量。但是这两个峰的m/z相加所得是目标分子量~是否存在这种可能[/color]
A5975MS 选中[同时SCAN/SIM]时,可以同时作SCAN、SIM,在使用此功能前是否要先编辑SIM方法(编辑好SIM检测参数)。请大家指教。谢谢!
最近在做一个糖苷类的检测方法,想用对硝基苯甲酸和糖苷上的羟基发生酯化反应显色后用紫外检测器检测,但是副产物太多,不能进行准确的定量,想请教下还有别的衍生化条件没? 我的衍生化条件是10mg糖苷样品+50mg对硝基苯甲酸加丙酮溶解后加1ml浓盐酸加热10min,条件是我自己摸索的,可能有很多的不规范的地方样品里可能有葡萄糖,蔗糖,葡萄糖甲苷,1-O-甲基-四苄基葡萄糖,1-羟基-四苄基葡萄糖
[em06] 四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。我让色谱公司按这个要求做了不锈钢柱子(柱填料:401有机载体(二乙烯基苯/乙基乙烯苯共聚体)60-80目),可是不出峰,后来把柱寄回去了,现在又寄给我的柱子(柱填料:10%PEG-20M CHROMOSORB PAW-DMCS 80-100目),峰是有了,可是分不开,我做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的氮气4圈,空气4.2圈,氢气4.5圈,后我又把氮气开到3圈,还是这个样子.是怎么回事呢,请高手赐教.谢谢!!!
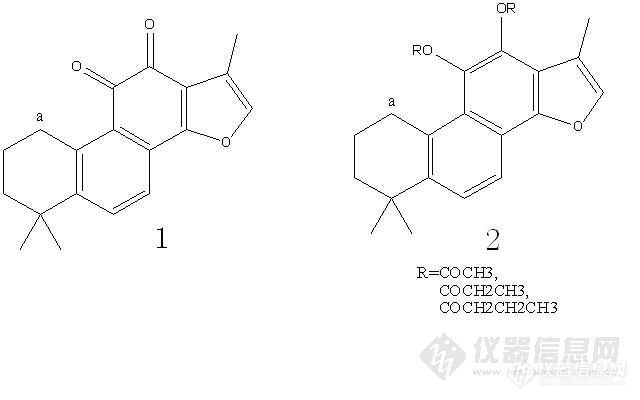
见附图,结构1反应后为结构2,同样是a位置的苄基氢(3.1-3.2ppm左右),在结构2却是一个矮矮的包,这是为什么呢?再附两张结构2不同R的图谱。[img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001261252_198822_1881730_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001261252_198823_1881730_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001261252_198824_1881730_3.jpg[/img]
最近在做兽药残留的液质方法,四环素类的做了四环素,土霉素,金霉素和强力霉素,四环素经基质加标后处理上机,四环素有40%左右已转化为差向四环素,不知大家有没遇到此类情况?我在提取时有超声10分钟/次×3次,感觉是因为超声的关系。有过相关经验的请麻烦指教
[size=3]撰写化学论文和编写其他化学式、公式较多的化学文档是一个很烦人的事,因为一会儿要编上角标,一会儿要编下角标,还要不断地在中文和西方之间切换,让你半天也写不出几页内容来。下面给出的word模板,将化学式、图片、公式等任何可以放在剪贴板中的内容瞬间替换文档中没有任何格式的文字、记号,你不必再为公式编辑和中西文切换耗时和烦恼。 动画是模板使用的操作演示,照葫芦画瓢吧。 操作说明: 1、这是一个word的模板文件,使用时直接运行它即可打开word窗口,其中页首有一个文本框和一个按钮,文本框输入文档中原来的内容(用中文或简单字母最方便),要替换的带格式的内容预先放在剪贴板中,例如,要将“硫酸”换成对应的化学式,你应该先在文档中编辑一个硫酸的化学式,然后复制到剪贴板。剩下的工作就是按下“替换剪贴板”的按键,OK 2、一般的word编辑软件(Office)考虑到安全性,默认设置是不允许其他宏运行的(我这个控件也算个宏吧),因此,此前你必须将“宏”安全设置成“中”级别,不然无法使用模板的。当打开模板时弹出可能有病毒的宏提示时,你不要谈“毒”色变,因为你自己的宏是已知的,毒从何来,人家只是善意提醒你而已。直接按下确定即可。 3、为了让操作方便,在操作中我增加了提取上下角标的图标放在工具栏上的操作,你如果不需要,就不必邯郸学步,其实,快捷键改变上下角标更方便,先选定要改下标的字符,同时按下“Ctrl”和“=”,就变下标了,要变上标,再多按一个键:Shift,即先按下“Ctrl”和“Shift”,再按下“=”(三键同按),就变上标了。这些快捷键若再按一次,又恢复到原来。 (复制:Ctrl+C;粘贴:Ctrl+V;撤消:Ctrl+Z;左对齐:Ctrl+L;右对齐:Ctrl+R;黑体:Ctrl+B;斜体:Ctrl+I) 4、你可以直接在打开的含控件的文档中录入和编辑文档,也可以将已经录入好的文档复制粘贴到这个模板的窗口中,然后,想替换什么就换什么。要改变结果的字号、字体、颜色等,请先设置好后再复制到剪贴板中。建议在录入文档时,用中文来代化学式、离子式,这样操作会快些。 5、这个模板中引入了两个控件,它们并不影响正常的录入和编辑,当你已经转化完全后,只要选定它们,将它们剪除就不妨碍文档的完整性了。 虽然已经将两个控件删除,但里面的代码没有删除,所以,你每次打开文件时可能都会弹出安全性提问,没关系的。本来可以将代码删除的,但考虑许多人不熟悉VBA,删了出问题,所以,我已经将代码窗口加密了。 如果大家对VBA在office中应用感兴趣,我可以做一个系列介绍,以回帖形式逐一发表介绍。 希望对你撰写化学文章有帮助。[flash=700,650]http://ng1.17img.cn/bbsfiles/images/2017/10/201008291201109598_01_0_3.swf[/flash][/size]
色谱缓冲液中加入四丁基溴化铵和四丁基硫酸氢铵有何作用,两种试剂有何区别
按国标14931.1做了一个畜禽肉中土霉素四环素金霉素的残留,标准上出峰顺序是土霉素、四环素、金霉素,我做出来的前两个峰没有分离出来,第三个峰出来了,不知原因。 流动相:乙腈/0.01mol/L磷酸二氢钠(用30%硝酸调PH2.5),35:65 柱子:ODS-C18 6.2×15 流速:1.0 波长:355我没有ODS-C18 6.2×15的柱子,用的是C18柱子因为着急认证,请知道的给予帮助。 新手啊 谢谢
用液相色谱做四环素的标液时,出现了土霉素的峰。本人同时做土霉素、四环素、金霉素和强力霉素。单标进样只有四环素中有土霉素的峰,而且土霉素峰还比四环素的高,其他标物单标均十分干净,包括土霉素单标,也未见杂峰。不是进样针头污染问题,也不是进样瓶污染的问题。难道是标物的问题?我同时做了盐酸四环素和液体四环素标物两种,均有土霉素的峰。请问到底是怎么回事?请大家给个意见。
[B]动物源性食品中四环素、沙星类[/B] 残留量的快速测定方法1 范围本方法规定了动物源性食品中四环素类、沙星类高效液相色谱的快速测定方法。本方法适用于动物源性食品中四环素类、沙星类高效液相色谱的快速测定。2.1 原理试样中的残留物经四环素类、沙星类快速检测前处理试剂盒处理,样液经四环素类专用层析柱净化、浓缩用高效液相色谱检测,外标法定量。2.2 试剂和材料除另有规定外,所有试剂均为分析纯,水为重蒸馏水。2.2.1 乙腈:色谱纯。2.2.2 甲醇:色谱纯。2.2.3 三乙胺(分析纯)2.2.4 磷酸(85%)(分析纯)2.2.5 磷酸氢二钠:优级纯。2.2.6 乙二胺四乙酸二钠。2.2.7 柠檬酸:分析纯。2.2.8 磷酸氢二钠溶液:0.2mol/L。称取28.41g磷酸氢二钠,用水溶解,定容至1000mL。2.2.9 柠檬酸溶液:0.1mol/L。称取21.01g柠檬酸,用水溶解,定溶至1000mL。2.2.10 Mcllvaine缓冲溶液:将1000mL0.1mol/L柠檬酸溶液与625mL0.2mol/L磷酸氢二钠溶液混合。2.2.11 Na2EDTA-Mcllvaine缓冲溶液:0.1mol/L。称取60.5g乙二胺四乙酸二钠放入1625mLMclllvaine缓冲溶液中,使其溶解,摇匀。2.2.12 标准品: 土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星纯度大于98 %。2.2.13 标准贮备溶液:分别称取土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星各10mg,用甲醇溶解并定溶于100 mL棕色容量瓶中,配制成100 µ g/mL的标准贮备液,置于-20℃保存,有效期三个月。2.2.14 混合标准工作溶液:用流动相稀释标准贮备溶液,配制成土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星均为10 µ g/mL的混合标准溶液。0~4℃避光保存。2.2.15 四环素、沙星类快速检测前处理试剂盒*。2.3 仪器和设备2.3.1 高效液相色谱仪:配紫外-可见光波长检测器。2.3.2 匀浆机。2.3.3 固相萃取机2.3.4 离心机:4000 r/min。2.3.5 调速多用振荡器。2.3.6 聚四氟乙烯离心管: 2.5 mL,50 mL,具塞。2.4 样品制备准确称取已捣碎的样品5.00 g于50 mL离心管中,先加入四环素、沙星类快速检测前处理试剂盒中的提取剂 (液体20mL),用调速多用振荡器150 rpm振荡3 min,,4000 r/min离心5 min,收集上清液10mL加入四环素专用层析柱中(使用前依次用5mL甲醇、5mL提取剂、5mL水激活)挤干,用2mL提取剂洗涤,用0.80mL甲醇洗脱,收集洗脱液,用0.2mL流动相定容至1.0mL。供仪器测定。2.5 测定2.5.1 液相色谱条件a) 色谱柱: C18柱,250 mm×4 mm(i.d.),粒度5µ m b) 流动相: 0.05 mol/L磷酸/三乙胺缓冲液(pH2.4)+乙腈(80+20,V/V) c) 流速: 1.0mL/min d) 柱温: 室温 e) 检测波长: 四环素类紫外检测器350 nm。沙星类 紫外检测器310nm;荧光检测器激发波长280nm,发射波长450nm。f) 进样量:50 uL。3.5.2 标准工作曲线绘制移取各移取四环素类、沙星类混和标准液,用流动相稀释成20 ng/mL、50 ng/mL、250 ng/mL、500 ng/mL标准工作溶液。按液相色谱条件(3.5.1)进行测定,以色谱峰的峰面积为纵坐标,与其对应的浓度为横坐标作图,绘制标准工作曲线,标准工作曲线范围:20.0~500 ng/mL。 3.5.3 试样测定 用微量进样器准确吸取试样溶液(3.4),按液相色谱条件(3.5.1)进行测定,记录色谱峰的保留时间和峰面积。3.6 结果计算按式(1)分别计算供试样品中的四环素类、沙星类残留量。 2×ci×Vω= …… (1)mω-水产品中四环素、沙星类残留量,μg/kg;ci -标准曲线上查出试样溶液中四环素、沙星类标准工作溶液的浓度,(μg/L);V-最终定容体积数,mL;2-换算常数;m-供试试料样品重量,g。本方法分别计算四环素类、沙星类结果。3.7 检测限本方法土霉素、四环素检测限为20µ g/kg;金霉素、强力霉素的检测限为50µ g/kg,沙星类为:5µ g/kg3.8 回收率 本方法土霉素、四环素、金霉素、强力霉素回收率为:75%~85%;沙星类回收率为:75%~85%相关谱图附件可见联 系 人:王 伦 手 机:13810239506 EMAIL:wwj613@sina.com
求助给位,我想问一下四丁基溴化铵、十六烷基三甲基溴化铵、苄基三甲基溴化铵怎么做红外,它们都很容易潮解的,吸水性很强,我问一下,做红外的时候样品怎么处理,或者这些物质的标准红外数据在哪可以查到啊
大家好!我用液相色谱法测定四环素,我们实验过程中回收很低总是在二三十左右,而且检测限也达不到方法的要求,请问是什么方面的问题,请大家支招,谢谢!
如何测富勒烯环内氢?谢谢[img]http://simg.instrument.com.cn/bbs/images/brow/em09511.gif[/img]
最近在做四环素类药物,开始液相做的,磷酸盐缓冲液流动相不行,某一个国标的,后来换草酸,出峰和响应均能达到要求,不过实际样品处理的时候回收率不好,前处理是1%高氯酸提取,重复,过C18小柱,也是国标处理方法,纯标准品过柱回收率还可以。液质目前问题是响应不好,仪器是安的6410,只能到50ppb,流动相是0.3%甲酸水和0.3%甲酸甲醇。做的好的达人请指点,多谢
目前要做四环素类及代谢的检测,看了很多文献,发现四环素类比较难检测。用的色谱柱也五花八门,不知道选哪一个了,下面是我看文献找到的柱子类型,请大侠指点。Waters Symmetry column C18 (150 ×2.1 mm, 3.5 μm)Xbridge C18 column, 2.1 × 150 mm id,5.0 μm particle sizeLuna C18 (150 × 2.0 mm, 3 m)Agilent EclipseXDB-C18 (150 mm ×2.1mm, 3.5 μm)waters T3ZORBAXSB-C18(100 mm×2. 1 mm 3. 5 μm)
联苯苄基氯(学名;4,4’-双氯甲基联苯)溶解方法?用ICP-AES测定其中的Ca,Fe,Zn,P,Na等元素(1--5ppm)。请高手指点样品处理方法为盼。谢谢啊http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
方法里头这个咪唑缓冲溶液:将68.08g咪唑、0.37g EDTA 和10.72g乙酸镁溶解在800mL水中,用乙酸调节pH7.2后,加水至1,000mL。这个EDTA是指乙二胺四乙酸还是乙二胺四乙酸钠含有乙二胺四乙酸的柠檬酸缓冲溶液:第1液:将21.0g柠檬酸溶解在水中至1,000mL。第2液:将71.6g磷酸氢二钠溶解在水中至1,000mL。在1.86g乙二胺四乙酸中加入307mL第1液和193mL第2液混合的溶液溶解。这个柠檬酸缓冲溶液里头的乙二胺四乙酸及时加热也很难溶解想问下做过这个方法的同行是怎么做的!!
1 别名英文名 四甲撑氧、氧杂环戊烷、四氢化氧杂茂、一氧五环、氧戊环;Tetrahydrofuran、Tetramethylene oxide. 2 用途 有机合成和制药生产中的溶剂、医药原料、合成橡胶原料、高能燃料。 3 制法 糠醛催化脱醛基,再催化加氢生成四氢呋喃。 4 理化性质 分子量:72.10 熔点: -108.5℃ 沸点:66℃ 液体密度(20℃):888kg/m3 折射率(20℃):1.4070 闪点: -14.4℃ 自燃点:321℃ 爆炸极限:2.0%~12% 在常温常压下四氢呋喃为具有乙醚气味的无色透明易燃有毒液体。在空气中能生成爆炸性过氧化物。其蒸气能与空气形成爆炸性混合物。遇明火、强氧化剂有引起燃烧的危险。与酸接触能发生反应。与氢氧化钠、氢氧化钾反应剧烈。与水、醇、酮、酯、醚、烃类等多数有机溶剂混溶。 5 毒性安全防护 最高容许浓度:200ppm(590mg/m3) 四氢呋喃有毒,有麻醉作用。蒸气可经呼吸道吸人,液体能经皮肤吸收而进人体内引起中毒。皮肤接触能引起灼伤和皮炎。轻度中毒可引起头晕、恶心、呕吐、气急、幻想、失眠、血压降低等症状。严重时可导致精神紊乱、昏迷等。 工作现场通风应良好,工作人员须穿戴个人防护用品,避免蒸气直接与人体接触。容器和生产设备应密封完好。 用镀锌铁桶包装。应贮存于阴凉、干燥、通风的库房中,要远离火种和热源,防止阳光直射。应与氧化剂、酸类物品隔离存放。搬运要轻装轻卸,严防包装破损。
现在用的是乙腈和水系统 要换成THF的系统 这个系统要怎么置换?乙腈—水—异丙醇—THF可以这样冲吗?如果要再换回来的话应该怎么置换?
大家有谁做过这个方法阿,蜂蜜中土霉素,四环素,金霉素和强力霉素残留量的测定方法(GB/T 18932.4-2002)。我按照国标做过,回收率很不理想。大家谁做过?请指点一下!多谢了
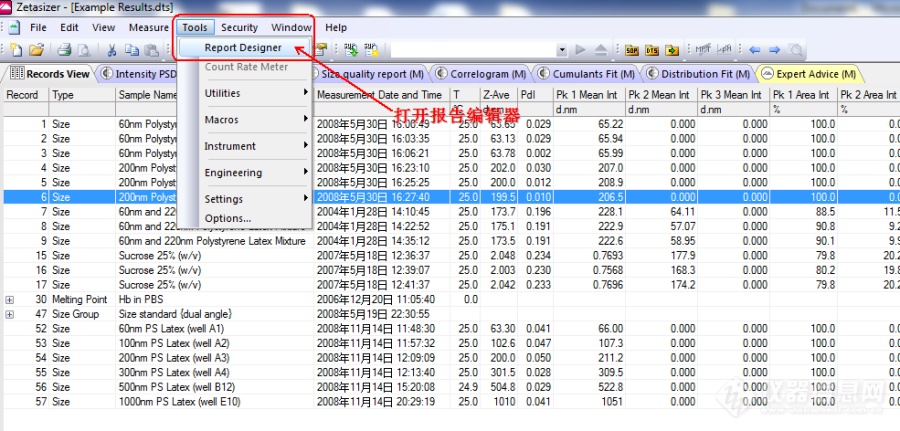
[align=center][size=20px]Zetasizer[/size][size=20px] [/size][size=20px]N[/size][size=20px]ano[/size][size=20px]软件报告[/size][size=20px]模板[/size][size=20px]的[/size][size=20px]编辑[/size][size=20px]和保存[/size][/align][align=center][/align][align=right][size=16px]作者:[/size][size=16px]MP[/size][size=16px]_Sherry[/size][/align][align=right][/align][align=left]马尔文帕纳科(原马尔文)Zetasizer Nano系列是非常受欢迎的纳米粒度电位仪,面世二十余年有广大的使用群体。该系列是利用动态光散射技术和电泳光散射技术高精度测量纳米级颗粒粒度及其Zeta电位的先进分析仪器。广泛应用于生命科学、生物制药、纳米材料、油漆、油墨和涂料、食品和饮料、给药系统及科学研究等需要分析颗粒或分子大小以及Zeta电位的应用领域。[/align][align=left]软件为客户设计了通用的报告模板,在日常分析过程中,可以根据实际的需要,方便地运用Nano软件来创建个性化的分析报告。下面将详细介绍如何编辑并且保存报告模板:[/align]1. 打开软件报告编辑器,选择主菜单上Tools-Report Designer[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546077634_9209_3895212_3.png[/img]2. 打开一个现有的报告模板,点击Open图标,跳出模板选择对话框,我们的模板文件必须保存在Malvern Instruments-Zetasizer Software下的Pages文件夹(具体的位置请在电脑中搜索此文件夹),按照所需的模板进行选择,以下为最常用的光强分布粒径报告模板Intensity PSD(M)为例。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546083257_2654_3895212_3.png[/img][/align][align=center][/align][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546081556_6199_3895212_3.png[/img][align=left]3. 在现有模板上进行修改。同一个模板有两种显示模式,Screen layout屏幕布局和Page Layout页面布局,前者是软件上该模板的显示内容,后者是打印报告时的显示内容,需要在哪个布局上显示修改,就在切换到哪个布局上进行编辑。[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546087381_7474_3895212_3.png[/img][/align][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546083872_5570_3895212_3.png[/img]对报告模板的修改通常是添加新的参数或者文本。[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546089815_3306_3895212_3.png[/img]4. 注意,修改完成后,将模板另存为新的模板文件,不要直接点击Save保存,标准模板是不允许直接修改保存的。保存时有两个地方需要输入新的模板名字,一是报告编辑器左上角文本框内;另一个是File-Save As另存为。 两个地方输入的名字要是相同的。[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546087198_1436_3895212_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546088350_9775_3895212_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546089573_9036_3895212_3.png[/img]5. 新模板的选择:将报告编辑器和Nano软件关闭,重新打开Nano软件,在Configure中Report Pages找到新存的模板名称,选中画勾,就能在Nano软件的报告选项卡上显示了。[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291546095194_2033_3895212_3.png[/img]需要看该报告的结果或者打印该页报告,就将选中的记录切换到该报告卡后查看或者打印。
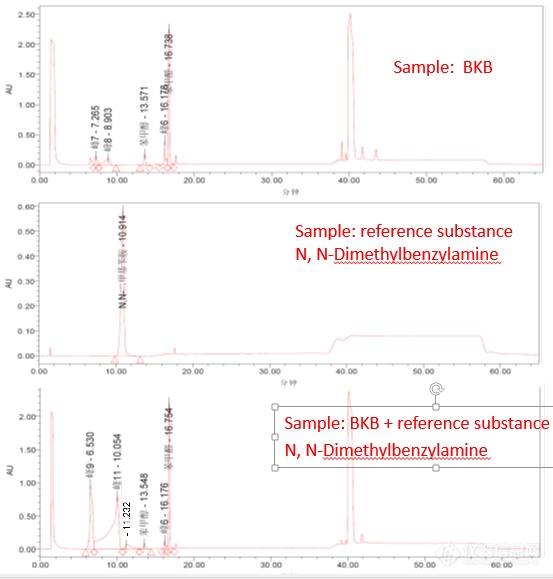
[color=#444444]最近在测苯扎溴胺(BKB)中苄基二甲基胺的残留,在测试条件下标样苄基二甲胺能够正常出峰,而进BKB样品时,苄基二甲基胺却不出峰,在BKB样品中加入苄基二甲基胺标样,却出现了两个奇怪的峰见色谱图,不知这是什么情况?[/color][color=#444444]色 谱 柱:端基封闭的C18(150×4.6mm,5μm)。[/color][color=#444444]柱 温:30℃[/color][color=#444444]流动相A:磷酸盐缓冲液(取己烷磺酸钠1.09g、磷酸二氢钠6.9g,溶于适量水中,用磷酸调节pH至3.5,用水稀释至1000ml,摇匀,即得。[/color][color=#444444]流动相B:甲醇[/color][color=#444444]时间(分) 流动相 A(V/V) 流动相 B(V/V)[/color][color=#444444]0-10 80 20[/color][color=#444444]10-14 80-50 20-50[/color][color=#444444]14-35 50 50[/color][color=#444444]35-36 50-20 50-80[/color][color=#444444]36-55 20 80[/color][color=#444444]流 量:1.0ml/min[/color][color=#444444]检 测 器:二极管阵列检测器(DAD)[/color][color=#444444][img=,553,579]https://ng1.17img.cn/bbsfiles/images/2019/06/201906031402122584_6731_1823055_3.png!w553x579.jpg[/img][/color]
四环素族配了贮备液,然后再稀释成中间混标1000ng/ml其中金霉素特别易降解 配置中甲醇做为容剂 。冷藏保存中间液。20几天就降解了。谁有好的办法,让四环素族降解的慢点。
今天做了一组pH对吸附四环素的影响,范围3到8,用紫外可见光度计测浓度,出来的结果我觉得不太对,pH在2、3时,测量值很高,远远超过了我初始溶液的浓度,我觉得很奇怪,这个结果肯定不对,但是不知道为什么。是pH会影响四环素的吸光度吗?在什么pH范围内测的浓度比较准确?请各位大佬指点迷惑。
土霉素、金霉素和四环素的检测方法 1. 分析目标化合物土霉素,金霉素,四环素2. 仪器设备带荧光检测器的高效液相色谱仪。3.试剂除下列试剂外,使用附录2所列试剂。咪唑:特级咪唑缓冲溶液:将68.08g咪唑、0.37g EDTA 和10.72g乙酸镁溶解在800mL水中,用乙酸调节pH7.2后,加水至1,000mL。含有乙二胺四乙酸的柠檬酸缓冲溶液:第1液:将21.0g柠檬酸溶解在水中至1,000mL。第2液:将71.6g磷酸氢二钠溶解在水中至1,000mL。在1.86g乙二胺四乙酸中加入307mL第1液和193mL第2液混合的溶液溶解。苯乙烯二乙烯苯共聚物小柱 (265mg):在内径8~9mm 聚乙烯管中填充265mg柱色谱用苯乙烯二乙烯苯共聚物或具有同等分离特性的物质。4.标准品盐酸土霉素:本品1.000mg 含有土霉素0.850mg以上效价,分解点为190℃~194℃。盐酸金霉素:本品1.000mg含有盐酸金霉素0.900mg以上效价,分解点为210℃以上。盐酸四环素:本品1.000mg 含有盐酸四环素0.900mg以上效力,分解点为214℃以上。5.试验溶液的制备a 提取方法①肌肉、肝脏和肾脏
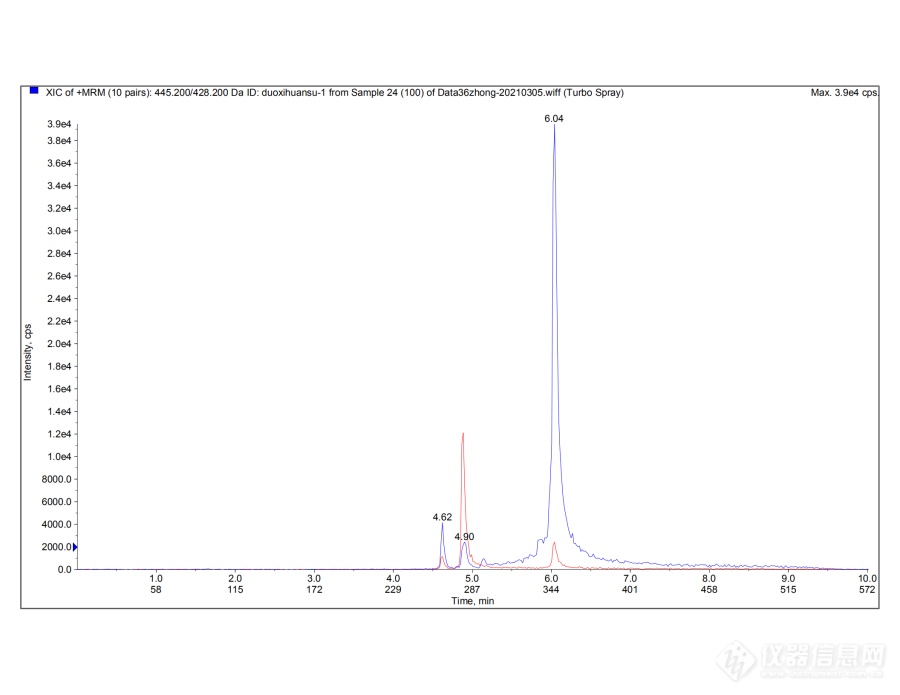
[img=,690,532]https://ng1.17img.cn/bbsfiles/images/2021/03/202103061051370228_8391_3101087_3.png!w690x532.jpg[/img][img=,690,532]https://ng1.17img.cn/bbsfiles/images/2021/03/202103061051370179_2403_3101087_3.png!w690x532.jpg[/img][img=,690,532]https://ng1.17img.cn/bbsfiles/images/2021/03/202103061051373177_4714_3101087_3.png!w690x532.jpg[/img][img=,690,532]https://ng1.17img.cn/bbsfiles/images/2021/03/202103061051371489_6174_3101087_3.png!w690x532.jpg[/img][img=,690,532]https://ng1.17img.cn/bbsfiles/images/2021/03/202103061051374251_5561_3101087_3.png!w690x532.jpg[/img]我现在在做(2015版)化妆品安全技术规范中36种抗感染类药物检测,其余物质各方面都还不错,昨天[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]进了四环素类中四环素、金霉素、土霉素、多西环素、米诺环素100ng/mL标样,各图谱所示物质见图片右上,流动相A:0.2%甲酸水溶液;B:0.1%甲酸甲醇溶液,溶剂为流动相A:B=1:1,进样量5μL。其中米诺环素出了双峰且响应很差,线性很差;四环素出双峰,金霉素出三个峰,多西环素有拖尾和前沿现象。有没有做过四环素类的老师指导一下,若能上传一下您做的四环素类的图谱参考一下感激不尽。标液是安谱买的36种物质混标。
THF的氢谱是两个多重峰,而碳谱是两个单峰,分子结构四个碳在同一平面,氧在平面外,有一个对称面,碳2、5和3、4等价,氢质子是怎么耦合的呢?每个亚甲基的两个质子应该不等价,同碳耦合是否存在?另外,看到文献报道苯并七元脂环的结构,七元环1,2位是苯,4位是氧,3,5位各有一个取代基,3、5氢有W耦合,4J=6。四氢呋喃也会有这种情况吗?4JW耦合的原因是什么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP