求:丙环唑加苯醚甲环唑混配制剂的 分析方法,谢谢大家 的帮助!!!
各有关单位及专家: 由吉林省农业委员会提出并归口,吉林农业大学、省参茸办公室等单位起草的《人参中丙环唑残留量的测定方法 GC-ECD法》、《人参中异菌脲残留量的测定方法 HPLC法》、《人参菌核净残留量的测定方法 GC-ECD法》、《人参中噻虫嗪的测定方法》、《无公害农产品 人参病虫害防治农药使用规范》、《人参生产基地建设要求》、《优质高产人参生产技术规程》等7项吉林省地方标准,已经形成地方标准征求意见稿。现在全省范围内公开征求意见,请有关单位、专家和社会各界积极参与,并于2011年12月22日前反馈具体的修改意见或建议。 修改意见或建议请采取回函的形式反馈到相应标准起草单位,具体联系方式如下: 吉林省农业委员会 地址:长春市人民大街1363号 邮政编码:130033 联系人:董广夏 电话/传真:88906017 手机:15164352281 电子邮箱:weichy@yeah.net 二〇一一年十一月十四日 附件 1: 丙环唑.rar 附件 2: 菌核净.rar 附件 3: 人参农药使用规则.rar 附件 4: 人参生产基地建设要求.rar 附件 5: 噻虫嗪.rar 附件 6: 异菌脲.rar 附件 7: 优质高产人参栽培技术规程.rar 附件 8:吉林省地方标准征求意见反馈表.doc
哪位老师用GB23200.121法测过呋虫胺、氰霜唑、环丙唑醇,定量限附近做不出来,比标准高了10倍
哪位老师用GB23200.121法测过??呋虫胺、氰霜唑、环丙唑醇,定量限附近做不出来,比标准高了10倍
环氧氯丙烷怎么做,好难做啊!!!我用吹扫捕集--质谱,要ppm级的才可检出,怎么才可以做到饮用水标准的0.4ppb啊。有人做这个项目的吗?你们都是怎么做的?
我采用的是甲醇:水96:4,或者甲醇:水75:25做流动相,检测波长223nm,测原药的时候没有问题,但是试样(250g/L丙环唑乳油)却总是分不开。[em0809] 我做液谱分析还太少,请达人帮忙分析分析
大连:开展全市核酸检测,暂停殡葬祭奠及婚姻颁证服务,全市人员非必要不离连,确需离开者需持检测阴性证明
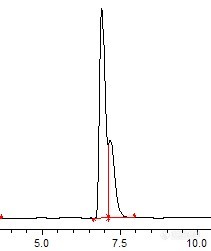
http://ng1.17img.cn/bbsfiles/images/2011/02/201102241706_279281_1928138_3.jpg我用的岛津的液相检测丙环唑,C18柱,流动相是甲醇+水=90:10,波长220nm,出的是双峰,但是连在一起,不知道有没有问题,不知道大家做是不是这样?
学校实验室的试剂一直有特定厂家订购,平常订试剂很方便,当天送过来,一年节一次帐。但是最近实验室订丙酮的时候,厂家说没有了,得等一周。师弟就在网上找厂家,但是说丙酮类这种试剂,如果采购的话需要去公安局办理证件。太麻烦啊,想问一下不办证件的话,怎么采购啊?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=174571]GB 23549-2009 丙环唑乳油[/url]
流动相是甲醇, 甲酸铵(2mmol/L)+0.01%甲酸水,但是环丙唑醇是两个峰,怎么回事呢?[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309191544148736_6337_5346633_3.png[/img]
美国输日红加仑子检测出丙环唑超标輸入時のモニタリング検査の結果、米国産生鮮レッドカラントから基準値を超えるプロピコナゾール(PROPICONAZOLE,用途:殺菌剤 基準値:0.05ppm)が検出されたと発表した。この結果を受け、米国産レッドカラントのモニタリング検査の頻度を30%に引き上げると通知。
环丙胺和正丙胺的沸点很接近,想要分离这两者应该用什么色谱柱,求大神指点
流动相是甲醇, 甲酸铵(2mmol/L)+0.01%甲酸水,但是环丙唑醇是两个峰,怎么回事呢?[img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309200708288399_5763_5346633_3.png[/img]
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]吹扫捕集做环氧氯丙烷不出峰
需要买丙酮等一些违禁品卖家要我们提供证书合理吗
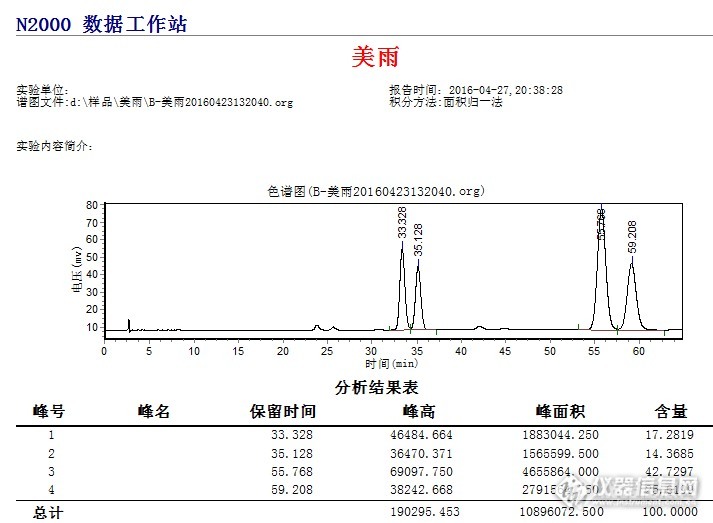
前段时间我在本网站偶然读到上海月旭科技有限公司的色谱柱试用的信息,就抱着试试的心理报了名。没想到过几天真的收到该该色寄来的新型色谱柱,我打开一看这么短,规格是Boltimate C18, 4.6*50mm,就没往心上去。后来月旭打电话过来介绍了柱子性能。我也仔细看来一下说明,就十分感兴趣。于是就开始做开了试验。没想到还真的分离效果可比25cm的普通C18柱,并且出峰时间快,这样就节省了时间。更为优秀的一点是节省了大量的流动相。分析一个样品,从成本、时间上降低不少。我用同样的流动相分析同一个样品上海月旭的短柱子节省时间为1/4。我分析的是复配制剂,丙环唑、苯醚甲环唑。用甲醇:水=50:50,在15分钟内很好的分离,而同样条件下,我以前的25cm柱子1小时还没出完。见下图。 http://ng1.17img.cn/bbsfiles/images/2016/04/201604272039_591677_1962166_3.png 图1 月旭核壳柱的色谱图----http://ng1.17img.cn/bbsfiles/images/2016/04/201604272059_591684_1962166_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/04/201604272059_591684_1962166_3.jpg 图2:月旭核壳柱产品照片---------http://ng1.17img.cn/bbsfiles/images/2016/04/201604272040_591679_1962166_3.png 图3 依利特 C18 5um,4.6*250柱色谱图-----------------如果用以前的25cm的柱子正常分析,流动相的有机相比例要达70~80%,且分析时间20多分钟, 月旭这个核壳柱分析效率可见一斑。应该说如果推广开来,他给使用单位带来的经济效益和分析效率是非常高的
我现在要检测环氧氯丙烷的水份,但不想用GB的GC法(没有TCD检测器),现用KF法检测时,甲醇会与样品反应造成检测结果偏高。我想请老师们指点一下,用KF做环氧氯丙烷的水份,最好用哪种溶剂好??????????
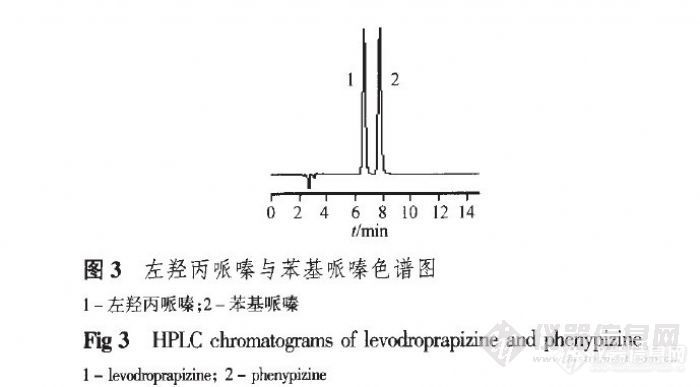
RP-HPLC测定左羟丙哌嗪缓释片的含量 鄢琳1,李铜铃H,张蓉琴1,许小红1,郑鹏程1,胡亚敏2(1.四川大学华西药学院临床药学教研室,l四川 成都610041;2.浙江大学第一附属医院药剂科,浙江杭州3100013)摘要:目的建立左羟丙哌嗪缓释片含量测定方法。方法采用反相高效液相色谱法,色谱柱为Diamonsil C18柱(4.0 mm×200mm,5um),流动相:甲醇.0.05 mol·L。1二乙胺水溶液(20:80),磷酸调pH至3.0,紫外检测波长:240 Bin。结果左羟丙哌嗪在5.03。50.30/xg·mL。内与峰面积呈良好线性关系,辅料对含量测定无干扰,平均回收率为99.93%,RSD为1.26%(n=9),方法精密度实验RSD=0.79%(n=6)。结论该法简便、准确、重复性好,适用于左羟丙哌嗪缓释片的含量测定。关键词:左羟丙哌嗪;反相高效液相色谱;缓释片;含量测定http://ng1.17img.cn/bbsfiles/images/2012/07/201207242117_379517_2432394_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207242117_379519_2432394_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207242118_379521_2432394_3.jpg
[color=#333333]环氧氯丙烷是用二氯甲烷做溶剂的,但是我没有,可以用二硫化碳代替吗?[/color]
请问一下买的丙酮中的环氧氯丙烷标准品,可以用丙酮做溶剂稀释吗,要用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]操作,标准里是用二氯甲烷做溶剂
刚做气相,做环氧氯丙烷的方法,结果不出峰,请教各位专家。情况如下安捷伦7890A,顶空进样,FID检测器顶空条件:传输温度142 ℃,炉温60℃,加热时间10min色谱条件:进样口150℃;柱温,初始40℃,保持4min,以30℃/min升至150℃,保持2min;检测器FID ,250℃。柱:安捷伦19091J-413 (HP-5)标准溶液:环氧氯丙烷用二氯甲烷配制,浓度497mg/L;取标液50微升加入盛有10.00mL纯水的顶空瓶中,密封,进样。结果发现只有溶剂峰二氯甲烷的峰,没有别的峰,怎么回事?
我现在用液质做tepa三-(1-氮杂环丙基)氧化膦,可是买不到标准品,大家咋买到的啊,谁有用液质做的色谱图啊!http://images.basechem.org/struct/2011-03/ff30a0213c0d51d45505574710dd5862.gif
请教各位:环氧氯丙烷用什么柱子做,效果好?我现在用的是FID检测器,柱子是强极性的INNOWAX柱,但响应不高。决定换柱子试试,不知FFAP柱如何?环氧氯丙烷这个项目好做吗?回收率一般如何呢?期待各位的意见和建议。
给一个铸造厂做环评验收,一开始说在河边可以办排污许可证,现在又通知说河边防护距离不够,不给排污许可证,这种情况怎么办
求助使用硅胶柱进行苯醚甲环唑原药顺反异构体分离,具体可用柱子型号有没有,正相很少做,怕买了做不好很麻烦。实验室设备都是安捷伦的液相,安捷伦工程师没有做过这个原药的方案。。。。推荐了一款大概可用的但是不敢买。
各位老师,做hj639全项,仪器条件和标准一样的,其他参数都是好着的,环氧氯丙烷不出峰,请问是什么原因

在做原料中环氧乙烷和环氧丙烷的检验方法,遇到了限度问题,想请教一下,一方面,在2020版药典四部,所有对环氧乙烷进行检验的方法里限度都是0.0001%(1ppm)另一方面,在ICH M7(R1)中的第26页,恰好以环氧乙烷为例演示了限度值是怎么计算的,就是以CPDB查询到的TD50来计算限度,最后计算结果是21.3微克/人/天,如下面的图,这样不就是对应原料限度是21.3ppm吗?所以,我应该以1ppm还是21.3ppm为限度呢?环氧丙烷按照ICH的方法,根据TD50计算是74.4ppm,药典四部有两个品种做环氧丙烷,限度分别是0.0005%和0.001%,环氧丙烷做的比较少。但是都比按TD50计算的结果小很多。欧洲药典中曲克芦丁有检环氧乙烷,限度也是1ppm,所以这个1ppm是出自哪里呢?是否应该按1ppm来作为限度呢?有帖子说05版药典残留溶剂附录中对环氧乙烷有限度规定,但现在的2020版或2015版中都没有查到,是否因为环氧乙烷是基因毒性杂质,所以没有在残留溶剂里面见到了,而ICH M7是对基因毒性杂质进行说明的。所以究竟以1ppm还是以21.3ppm做限度呢?[img=,650,899]https://ng1.17img.cn/bbsfiles/images/2021/03/202103081021496056_5338_2789522_3.jpg!w650x899.jpg[/img]
各位大侠,我测量环氧丙烷用的内标物是甲基叔丁基醚,样品里还有大量甲醇和水.没出来的环氧丙烷值比实际值偏大.我配了一份已知质量的环氧丙烷,验证标准曲线没问题,但往其中加水稀释后,测量值大大偏高.请问这是什么原因???按理说内标法不会出现这种情况呀?稀释时连同内标物一同稀释了呀!!!
根据GB 29704-2013 检测环丙氨嗪及三聚氰胺,流动相由乙酸铵改成了0.1%甲酸水,用乙腈配的标样 峰能走出来,用空白基质配的标样基质效应很强,经过前处理后的加标样品基本做不出来,是为什么???求大神赐教!![img]https://simg.instrument.com.cn/bbs/images/brow/em58.gif[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP