请教各位大侠:氢氟酸是否是强氧化性酸?能否与聚苯脂发生反应?聚苯脂就是聚对羟基苯甲酸苯脂??急用,在线等。我查了很多资料都没有相关的结论。
[table][tr][td][table][tr][td]用waters Uplc 、waters T3柱子,以95%乙酸铵、5%甲醇为流动相,检测糕点样品中5种防腐剂(前处理过程按国标),发现柱效降低飞快,新柱子跑不到一百个样品,每个峰都出现很大的肩峰,原来分离良好的糖精钠与脱氢乙酸无法分离;柱子用水、甲醇、乙腈、异丙醇混合清洗后在前十个左右样品状况良好,之后 的样品再次出现肩峰、糖精钠与脱氢乙酸无法分离现象。这些现象原来在HPLC上并不明显。请问大家用什么柱子配合UPLC做防腐剂检测?如何防止柱效快速下降?[/td][/tr][/table][/td][/tr][tr][td] [/td][/tr][/table]
1.将耐氢氟酸电极连接到ph计的输入端,正确连接。2. 正确配制耐氢氟酸电极浸泡混合液:取37.25克氯化钾和1.75克混合磷酸盐粉末配制成250ml混合溶液。3.严禁用耐氢氟酸电极测量能溶解PVC以及ABS的有机溶液,该体系对电极造成永久性破坏。4.耐氢氟酸电极导线及绝缘部分要保持清洁干燥,每次使用后将耐氢氟酸电极用纯水清洗干净,并放入装有混合溶液护套内或混合溶液中浸泡。5.特别注意:定位时,使用PH6.86和PH9.18的缓冲溶液,不可使用PH4.0的邻苯二甲酸氢钾缓冲溶液,若不慎使用,立即用混合溶液浸泡至性能恢复。6.强氧化性体系及高亲脂物质含量极大的体系长期接触会对耐氢氟酸电极造成损害,短期接触后对电极性能有不良影响,出现此状况,立即用混合溶液浸泡至性能恢复。7.耐氢氟酸电极使用时先用PH6.86缓冲溶液中浸泡10分钟,实验室电极使用后可长期浸泡在混合溶液中,工业耐氢氟酸电极使用后必须再次校准时,并且在混合溶液中浸泡4小时以上,使参比电极和工作电极有一个稳定的扩散电位,使用时轻甩耐氢氟酸电极以确保内充液与工作膜有效接触。
求助各位:氢氟酸可以用pH计测量吗?或者有什么其他的办法测量氢氟酸的pH值?
氢氟酸对玻璃有腐蚀作用,那如何取用准确体积的氢氟酸呀。可以用普通的玻璃材质的移液管或量筒吗。
慕尼黑上海分析生化展,迪马科技最新推出了Inspire 苯基系列色谱柱,一直忙未给大家及时普及,今天来说说第一款Inspire PFP ( 五氟代苯基) 是Inspire 液相色谱柱家族新成员,针对分离极性化合物过程中的保留时间和分离度问题而特别设计。Inspire PFP 凭借其优异的选择性,可为极性化合物、复杂天然产物、位置异构体和其它相关化合物在C18 和C8 色谱柱上的分离提供一个替代和补充。Inspire PFP 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式,并具有多种作用机理,因而能够同时分离检测不同极性化合物的混合物,为目前难以解决的复杂极性和亲水性样品的分离分析提供了强有力的工具,可轻松解决其它色谱柱面临的分离难题,为用户实现强极性分析物的优异选择性提供一种更加便捷的途径。同时也为色谱工作者使用简单流动相,避免使用极端pH 条件和准备复杂流动相提供了可能性。Inspire PFP 色谱柱特点• 五氟代苯基硅烷键合在高纯硅胶基质上• 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式• 对极性化合物具有独特的保留能力• 良好的峰形、超高的柱效、分离度和使用寿命• 适用于芳环类化合物或长共轭体系化合物的分离• 优异的批次重现性增强位置异构体分离能力官能团位置的微小差异可以极大的影响分子性能,在许多情况下,传统的C18色谱柱根本无法扑捉到这种细微的差异。然而,Inspire PFP的多功能选择性却可以区分由于分析物内部微小位置变化而导致的分析物的空间位阻变化还是分析物的偶极矩偏移。色谱柱如图所示 规格 150 × 4.6 mm, 5 μm 流动相0.1% 甲酸乙腈溶液:0.1% 甲酸水溶液 = 40:60 流速1.5 mL/min 柱温室温 检测器 UV 254 nm 样品1. 3,4-二甲氧基苯酚 2. 2,6-二甲氧基苯酚3. 3,5-二甲氧基苯酚4. 2,6-二氟苯酚5. 2,4-二氟苯酚6. 2,3-二氟苯酚7. 3,4-二氟苯酚8.3,5-二甲基苯酚9.2,6-二甲基苯酚10.4-氯-3-甲基苯酚11.4-氯-2-甲基苯酚12.3,4-二氯苯酚13.3,5-二氯苯酚http://www.dikma.com.cn/u/image/2016/09/06/1473147613188048.jpg苯氧酸类化合物分子上卤素的加入可以从根本上增强化合物的极性,而极性的变化通常伴随着反相色谱柱在保留时间和分离能力上困难的增加。此时使用InspireTM PFP 是解决保留问题的最有效的方法。InspireTM PFP利用偶极-偶极和氢键作用更好地保留,区分和分离极性卤化化合物。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:0.1% 甲酸水溶液 = 50:50流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 苯氧乙酸2. 邻氯苯氧乙酸3. 对氯苯氧乙酸4. 2,4-二氯苯氧乙酸5. 2,4,5-三氯苯氧乙酸6. 2,4,5-三氯苯氧丙酸http://www.dikma.com.cn/u/image/2016/09/06/1473147817102957.jpg类固醇通过整合偶极-偶极、π-π和氢键机理,InspireTM PFP实现标准反相条件下极性化合物的最佳分离。色谱柱 如图所示 规格 150 × 4.6 mm, 5 μm 流动相甲醇:水 = 60:40 流速1.5 mL/min 柱温室温 检测器UV 254 nm 样品1.泼尼松龙3.地塞米松5.氢化可的松21-乙酸酯7.可的松-21-乙酸酯2.泼尼松4.皮质酮6.11-α羟孕酮8.11-酮孕甾酮http://www.dikma.com.cn/u/image/2016/09/06/1473148006619700.jpg甲基苯乙酮异构体目标分析物上的基团位置变化可以影响化合物的偶极矩,这种变化可以很容易被高电负性的氟原子和其它保留机理察觉,因此InpireTM PFP可以有效地用于分离甲基苯乙酮的位置异构体。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相甲醇:水 = 50:50流速1.0 mL/min柱温室温检测器UV 254 nm样品1. 邻 -甲基苯乙酮2. 对 -甲基苯乙酮3. 间 -甲基苯乙酮http://www.dikma.com.cn/u/image/2016/09/06/1473148212903667.jpg核苷酸和核苷色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相0.1% 甲酸水溶液流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 胞嘧啶2. 5'-CMP3. 5'-UMP4. 5'-GMP5. 尿苷6. 胸腺嘧啶 http://www.dikma.com.cn/u/image/2016/09/06/1473148405914511.jpg抗胃酸药色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:20 mM 磷酸氢二钾(pH 7.0) = 20:80流速1.0 mL/min柱温室温检测器UV 220 nm样品1.法莫替丁2.西咪替丁3.尼扎替丁4.雷尼替丁 http://www.dikma.com.cn/u/image/2016/09/06/1473148597658988.jpg氧化应激标记物色谱柱 如图所示规格 150 × 4.6 mm, 5 μm[/
[font=&]1.将耐氢氟酸电极连接到ph计的输入端,正确连接。[/font][font=&]2. 正确配制耐氢氟酸电极浸泡混合液:取37.25克氯化钾和1.75克混合磷酸盐粉末配制成250ml混合溶液。[/font][font=&]3.严禁用耐氢氟酸电极测量能溶解PVC以及ABS的有机溶液,该体系对电极造成永久性破坏。[/font][font=&]4.耐氢氟酸电极导线及绝缘部分要保持清洁干燥,每次使用后将耐氢氟酸电极用纯水清洗干净,并放入装有混合溶液护套内或混合溶液中浸泡。[/font][font=&]5.特别注意:定位时,使用PH6.86和PH9.18的缓冲溶液,不可使用PH4.0的邻苯二甲酸氢钾缓冲溶液,若不慎使用,立即用混合溶液浸泡至性能恢复。[/font][font=&]6.强氧化性体系及高亲脂物质含量极大的体系长期接触会对耐氢氟酸电极造成损害,短期接触后对电极性能有不良影响,出现此状况,立即用混合溶液浸泡至性能恢复。[/font][font=&]7.耐氢氟酸电极使用时先用PH6.86缓冲溶液中浸泡10分钟,实验室电极使用后可长期浸泡在混合溶液中,工业耐氢氟酸电极使用后必须再次校准时,并且在混合溶液中浸泡4小时以上,使参比电极和工作电极有一个稳定的扩散电位,使用时轻甩耐氢氟酸电极以确保内充液与工作膜有效接触。[/font]
最近做二氧化硫曲线,线性不好查阅了论坛里的相关帖子,想购买0.2%盐酸副玫瑰苯胺溶液看了有推荐天津市化学试剂研究所的网络查询了没有购买方式希望使用了的同行们给推荐一下购买方式,多谢了之前使用的是FMP级的盐酸副玫瑰苯胺试剂配制的
客户做枸橼酸苯海拉明,用氰基柱做有关物质(按照药典上盐酸苯海拉明的方法),刚开始实验效果很好。用了两天后出现主峰分叉现象,换用之前一根C18(柱效不高)的色谱柱做没有分叉现象。还请大家多多指点,不胜感激!
[em06] 金属消化前处理的部分,由于ICP不具有防氢氟酸设备之前有很多人讨论过利用硼酸对轻氟酸做赶酸处里,但是还是有不太了解的地方想请问各位高手,如果每次消化加入4ml 的氢氟酸(40%),要加入多少量的饱和硼酸溶液才够用呢??以及氢氟酸与硼酸的反应方程序是如何呢?因为查过USEPA 3052方法里只有提到加入适量的硼酸,并没有明确的加入的剂量如果有哪位前辈知道希望提供噜还有是否有相关的文献是在探讨氢氟酸加入硼酸赶酸的探讨呢???不好意思ㄧ下子问了好多问题希望能得到解答喔 谢谢
消解方法要使用氢氟酸,氢氟酸对仪器的损害比较大,所以要尽量将之赶尽,听说加入硼酸比较好。问题是加入硼酸的浓度、体积、如何判断氢氟酸是否去除赶尽?硼酸加入过量又有什么影响?
请问哪位版友有农产品中三氯异氰尿酸、三乙膦酸铝、氟苯脲、氟吡甲禾灵、氟酰胺、环酰菌胺的测定方法? 三氯异氰尿酸在棉花、水稻上的测定方法,三乙膦酸铝在蔬菜、水果中的测定方法,氟苯脲在蔬菜、水果中的测定方法,氟吡甲禾灵在水果、咖啡豆中的测定方法,氟酰胺在稻米中测定方法,环酰菌胺在蔬菜、水果及其干制品中的测定方法。最好是国标或行标。先致谢了。
用邻苯二甲酸氢钾标定的氢氧化钠,算其对于硅氟酸的滴定度,应该怎样计算,谢谢!
用2,4-二硝基氟苯衍生测定氨基酸,可以把乙腈换成甲醇吗
以下是氢氟酸的标签内容:500毫升批号四赫有毒品厂址:太和路1004号公司:大渡河路188号上海化学世纪有限公司氢氟酸Hydrofluoric acid分子式:HF分子量:20.01符合:GB/T 620-1993试剂生产许可证号:xk13-2010279-176 I危规号:91035技术条件含量……………………………………≥40%灼烧残渣………………………………≤0.001%氯化物(Cl)…………………………≤0.0005%硫酸盐和亚硫酸盐(以SO4计)……≤0.001%磷酸盐(PO4)…………………………≤0.0001%氟硅酸盐(SiF6)……………………≤0.002%铁(Fe)………………………………0.00005%重金属(以Pb计)……………………≤0.0001%忧级纯 GR===============做土壤中Cu的时候,消解液是HNO3+H2O2+HF,发现试剂空白很高。将这个氢氟酸试剂直接进火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url],发现Cu元素的值有2.06mg/L我想请教一下,这样对照这个氢氟酸试剂的标签,这个氢氟酸合格么?
有没有那种水果或蔬菜本身含有苯甲酸和山梨酸之类的防腐剂?望高手指点,谢谢
如题,gcms要做校准,今天按照检定规程来设置参数,3个样品出来的谱图都是一个斜坡状的,各提取特征离子,均没找到,柱子是强极性的,极限温度是280,30m长。接着降低进样口温度为200,传输线250,降低柱箱初温,出来的谱图都是几个 ’几‘字型,各提取特征离子,八氟萘和硬脂酸甲酯没找到峰,六氯苯找到几个连在一起的不规则的峰,但最高丰度的特征离子碎片是88,不是284.无论我如何设置升温程序,任何时间点的最高离子碎片的质荷比都是88,当然溶剂峰除外。是不是我的柱子有问题?
因为氢氟酸有溶解玻璃的特性,普通的玻璃量器都不能量取氢氟酸,做氢氟酸试验时大家是如何量取和转移氢氟酸的?
[size=3][b]滴定 硫酸 硝酸 溶液 镁离子[/b][/size] 若混合酸溶液中含有氢氟酸、硝酸、硫酸,怎样分别测出其中各酸的含量,最好是简单的方法,像滴定的方法,另有溶液中含有铝离子、镁离子、硫酸,怎样测出其酸的含量以及铝、镁离子的含量,急求。
用高效液相色谱分析:邻氯苯基环戊酮; 2-吡啶甲醛;方酸; 2,5-二氧基-4-碘苯乙胺; 苯瞵酸; 戊亚酮胺的含量的检测条件,恳请各位专家帮帮忙啊!
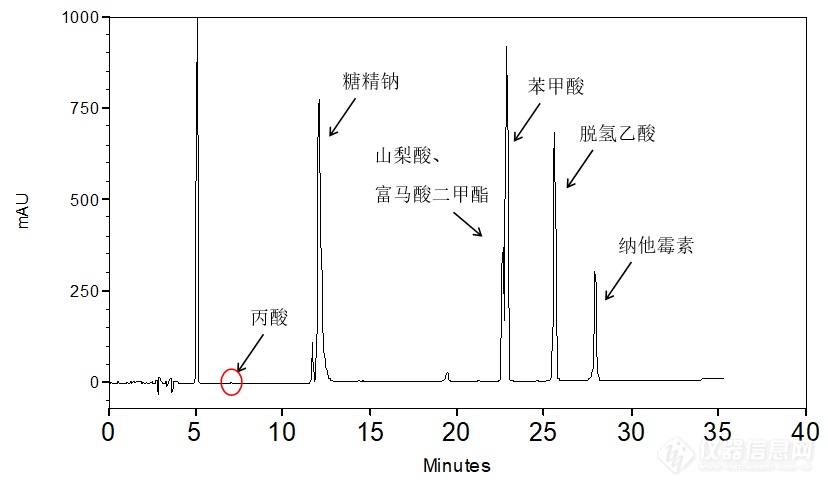
[align=center][b]食品中7种添加剂的共同分析——丙酸钠、山梨酸、脱氢乙酸、苯甲酸、糖精钠、纳他霉素和富马酸二甲酯[/b][/align][img=,99,45]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143105100_953_2222981_3.gif!w99x45.jpg[/img] [img=,121,46]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143267296_3413_2222981_3.gif!w121x46.jpg[/img] [img=,98,83]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143361658_9073_2222981_3.gif!w98x83.jpg[/img] [img=,89,50]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201143485158_3959_2222981_3.gif!w89x50.jpg[/img] 丙酸钠 山梨酸 脱氢乙酸 苯甲酸[img=,112,150]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144042246_934_2222981_3.gif!w112x150.jpg[/img] [img=,270,166]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144144504_7297_2222981_3.gif!w270x166.jpg[/img] [img=,123,58]http://ng1.17img.cn/bbsfiles/images/2018/04/201804201144263678_2065_2222981_3.gif!w123x58.jpg[/img] 糖精钠 纳他霉素 富马酸二甲酯丙酸钠、山梨酸、脱氢乙酸、苯甲酸、糖精钠、纳他霉素和富马酸二甲酯为食品中常见的添加剂分析项目,客户希望通过一种方法来同时实现7种添加剂的分析,且整体分析时间要求尽量短,以提高工作效率。首先,使用大曹三耀(原资生堂)CAPCELL PAK系列色谱柱中的第一选择色谱柱——通用型反相柱[color=#3366ff][b]CAPCELL PAK C18 MGII[/b][/color]来进行方法初筛;考虑到7种添加剂中的酸性化合物较多,为取得良好保留,在流动相的选择方面,首先尝试在酸性条件下(0.1%磷酸)进行分析,结果如图1所示。在酸性条件下,苯甲酸、山梨酸和富马酸二甲酯三者间分离情况不佳。[align=center][img=,690,405]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104146524_289_2222981_3.png!w690x405.jpg[/img][/align][align=center]图1 酸性条件下分析结果[/align][img=,431,219]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104139552_9327_2222981_3.png!w431x219.jpg[/img]在此条件下,进一步对梯度条件进行调整,并将流动相中的乙腈更换为甲醇,更换不同种类色谱柱,但均未能获得良好结果。接下来我们将流动相pH改变,将0.1%磷酸溶液更换为20 mmol/L磷酸二氢铵溶液进行分析,结果如图2所示。能够看到7个较明显物质峰得到分离。[align=center][img=,690,359]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104574536_4474_2222981_3.png!w690x359.jpg[/img][/align][align=center]图2 磷酸二氢铵条件分析结果[/align][img=,450,220]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191104576746_74_2222981_3.png!w450x220.jpg[/img]由于客户想要在短时间内得到良好分析,因此我们进一步将色谱柱由常规规格[b]S5 4.6 mm i.d. × 250 mm[/b]更换为[b][color=#3366ff]S3 4.6 mm i.d. × 100 mm[/color][/b],分析结果如图3所示。[align=center][img=,690,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191105363445_4814_2222981_3.png!w690x418.jpg[/img][/align][align=center]图3 MGII小粒径短柱分析结果[/align][img=,459,221]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191105376737_3055_2222981_3.png!w459x221.jpg[/img]但同时在分析中发现丙酸钠单标无法得到良好响应(图4),且富马酸二甲酯出峰行为异常(图5)。[align=center][img=,690,337]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191106365666_7879_2222981_3.png!w690x337.jpg[/img][/align][align=center]图4 丙酸钠单标分析结果[/align][align=center] [/align]如图5结果所示,将7种添加剂各取100 μL后稀释到1 mL配制混合标准品时(第一次混合样品,终浓度为各0.1 mg/mL),未见富马酸二甲酯峰出现,此时再将此溶液与富马酸二甲酯单标以4:1比例混合后(第二次混合样品,富马酸二甲酯终浓度0.28 mg/mL,其他为0.08 mg/mL),富马酸二甲酯峰出现,同时2号峰峰面积增大。富马酸二甲酯单标进样无异常。因此富马酸二甲酯在混合溶液中可能存在[color=red]降解反应、溶解度不佳或其它原因[/color],导致出峰行为异常,建议对该物质性质进行考察,或进行方法学验证。[align=center][img=,690,450]http://ng1.17img.cn/bbsfiles/images/2018/04/201804191106370947_4629_2222981_3.png!w690x450.jpg[/img][/align][align=center]图5 富马酸二甲酯分析结果[/align][align=left] 综上所述,使用[b][color=#ff0000]CAPCELL PAK C[sub]18[/sub] MGII S3 4.6 mmi.d. × 100 mm[/color][/b]色谱柱可在[color=#3366ff][b]20 mmol/L磷酸二氢铵-乙腈[/b][/color]条件下基本实现7种常见食品添加剂的分析,但丙酸和富马酸二甲酯出峰异常,建议进一步进行方法学验证。[/align][align=left][/align][align=left] [/align][align=right] [/align][b][/b][align=right][b][color=#333333]LC Application Lab, Sanyofine China[/color][/b][/align][color=#333333][/color][align=right]Beijing, China[/align][color=#333333][/color][align=right]Phone 400 801 3103[/align]
在GCMS仪器检定、期间核查的时候,经常采用八氟萘验证信噪比;采用六氯苯测量重复性情况;而使用硬脂酸甲酯分析质量数准确性与谱库的匹配度。---------------------------------试问:为啥偏爱此三种物质?---------------------------------个人感觉:1.首先这三种物质均能检测到分子离子峰 2. 八氟萘与六氯苯均是苯环结构,比较稳定 3. 硬脂酸甲酯除分子离子峰外,在50-300u区间内产生比较稳定的碎片离子,且比例也相当。个人猜测推断,不知是否正确?欢迎大家指点一二,相互探讨下。。。
异氰基乙酸乙酯和N,N-二甲基苯胺是否可以用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?有没有方法条件呢?
[size=3][b]滴定 硫酸 硝酸 溶液 镁离子[/b][/size] 若混合酸溶液中含有氢氟酸、硝酸、硫酸,怎样分别测出其中各酸的含量,最好是简单的方法,像滴定的方法,另有溶液中含有铝离子、镁离子、硫酸,怎样测出其酸的含量以及铝、镁离子的含量,急求。
我们用盐酸福玫瑰苯胺法测食品中的亚硫酸盐遇到了问题,请大家帮帮忙。我们按国标配的曲线为什么颜色都一样啊,没有色阶,配了很多遍盐酸福玫瑰苯胺都不能解决。
请问大家阿有盐酸付玫瑰苯胺的配制方法啊?我做空气中的二氧化硫,按照标准配出来的颜色是紫红色的也,怎么不褪色?真是急死人
请问在哪里能购到检测二氧化硫的显色剂 盐酸付玫瑰苯胺?偶是湖北的,最好能在湖北境内买到.[em61]
我现在用付品红替代,但该试剂纯度很差,染料级的.请问做SO2的盐酸付玫瑰苯胺哪能买到
求有2-(4-羟基苯氧基)丙酸工业化技术的朋友吗?我想要中控分析(请不要留联系电话,可以常来看回复或者站短,谢谢支持!)
[color=#444444]顺酐.富马酸.六氢苯酐,用液相色谱如何分离,用什么流动相比较合适?[/color][color=#444444]哪位大侠知道,求指点[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP