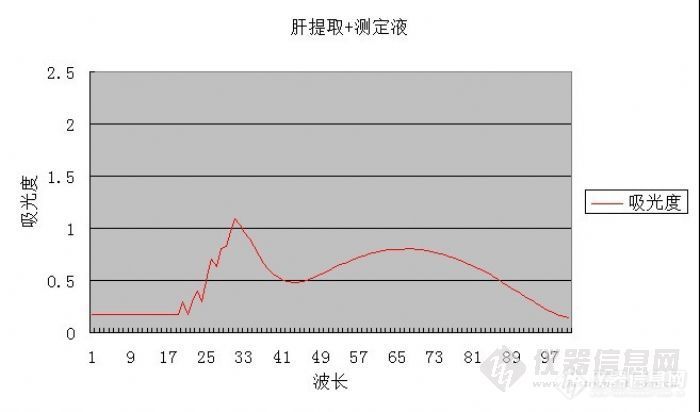
[size=4] 大家好,我的实验过程中遇到了些问题希望能得到大家的帮助,谢谢啊! 为了测定实验动物小鼠的肝肾中的[color=#940000]顺乌头酸酶[/color],我用提取液匀浆,[color=#940000]提取液[/color]成分(50mM Tris-HCl,1mM半胱氨酸,1mM柠檬酸钠,0.5mM MnCl2 ,PH8.0 ),离心后上清液与测定液(50mM Tris-HCl,20mM柠檬酸钠,PH8.0)反应,测紫外吸收,文献报道是240nm会有最大吸收,但是我的实验结果是在230nm有吸收峰,所以怀疑此230nm吸收峰是不是就是我要测的吸收峰,然后用排除法,因为测定液中的柠檬酸钠是酶反应的底物,当测定液中不加人底物,如果没有发现吸收峰的话那此吸收峰可能就是我要测的顺乌头酸酶跟底物柠檬酸钠反应产生的顺乌头酸的吸收,但是,紫外200-300nm扫描发现在200-235nm间有馒头状高吸收,是有底物条件下吸收峰的3倍甚至更高,这是为什么呢? 为什么我加入底物之后使紫外吸收显示出一个吸收峰,整体的吸光度下降那么多呢?[img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003312114_209275_2024887_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003312117_209276_2024887_3.jpg[/img][/size]
http://ng1.17img.cn/bbsfiles/images/2014/02/201402201500_490669_1060668_3.bmp问题1:如果基线出现较大波动,是否四氢呋喃氧化造成的问题2:流动相加冰醋酸后,PH为4.7,但是没出峰,是否还需要调节PH测乌头碱的条件如下:以乙腈-四氢呋喃(25:15)为流动相A,以0.1mol/L醋酸铵溶液(每1000ml加冰醋酸0.5ml)为流动相B,按下表中的规定进行梯度洗脱;检测波长为235nm。时间(分钟)流动相A(%)流动相B(%)0~4815→2685→7448~4926→3574→6549~58356558~6535→1565→85对照品溶液的制备取乌头碱对照品、次乌头碱对照品、新乌头碱对照品适量,精密称定,加异丙醇-三氯甲烷(1:1)混合溶液分别制成每1ml含乌头碱50μg、次乌头碱和新乌头碱各0.15mg的混合溶液,即得。
急需苯甲酰乌头原碱、苯甲酰次乌头原碱、苯甲酰新乌头原碱,哪位老师能提供,我高价购买。
求助,请大家帮帮忙啊~~~~本人在做乌头碱的实验,将乌头碱对照品在酸性条件下进行加热回流2h,用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]进行分析得一级质谱:618 二级质谱:568 三级质谱:536想知道从646到618 脱去的是什么基团,C8位有可能脱去羰基吗?还是脱去其他基团?以及三级质谱裂解的方式,求各位大侠帮助~~~~~谢谢啦~~~~
求助各位大侠:HPLC:乌头碱、次乌头碱、新乌头碱分离时乌头碱、次乌头碱始终分不开,二者只显示一个峰,无论怎么拉长时间都没有变化,这是怎么回事?条件:A:0.1%三乙胺+0.1%醋酸 B:乙腈 谢谢!
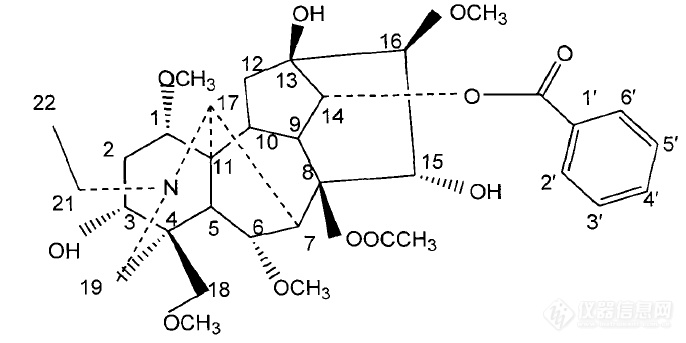
[color=#444444]乌头碱(Aconitine),很常见的一个化合物,结构式如下图,分子量也知道,现在需要一张质谱图片,HR-MS或者ESI-MS都可以的,或者哪篇文献或者毕业论文上有质谱谱图,麻烦把论文题目告诉我,谢谢![/color][color=#444444][img=,686,343]https://ng1.17img.cn/bbsfiles/images/2019/09/201909121123493821_9132_1823055_3.png!w686x343.jpg[/img][/color]
根据香港食物安全中心最新通报,昨日检测的15个水产样本中,一个捕捞的乌头鱼样本检出微量碘-131(每公斤7.7贝可)。 此含量没有超出食品法典委员会的指引限值(每公斤100贝可)。根据风险评估,正常食用该低辐射量的乌头鱼不会对健康构成风险。香港渔护署会继续密切监察情况。 在昨天的检测中,香港食物环境卫生署食物安全中心共检测了275个从日本进口的食品样本(包括31个空运样本和244个海路样本),当中有水产、蔬菜、水果、肉类、肉类制品、饮品及其他,检测的样本全部合格。 从三月二十二日至五月二十七日正午,香港食物环境卫生署食物安全中心已检测的渔产品样本总数有743个,包括七个乌头样本,测试结果全部合格。
美国权威消费杂志《消费者报道》(ConsumerReports)发布的调查报告指出,去年美国民众花在购买健康食品的费用累计高达267亿美元,但消费者却不知道贩售这些健康食品的业者其实并不能证实这些产品确实安全有效。《消费者报道》指出,包括乌头草在内的12种草药制成的保健食品可能对人体造成不良影响,消费者应避免服用。 美国权威消费杂志《消费者报道》(ConsumerReports)发布的调查报告指出,去年美国民众花在购买健康食品的费用累计高达267亿美元,但消费者却不知道贩售这些健康食品的业者其实并不能证实这些产品确实安全有效。由于缺乏具体规范,有些产品甚至含重金属、杀虫剂与处方类药品。《消费者报道》指出,包括乌头草在内的12种草药制成的保健食品可能对人体造成不良影响,消费者应避免服用。 这12种保健食品原料包括:乌头草(Aconite)、苦橙(BitterOrange)、榭树(Chaparral)、胶体银(ColloidalSilver)、款冬(Coltsfoot)、紫草(Comfrey)、紫罗兰(CountryMallow)、锗(Germanium)、白屈菜(GreaterCelandine)、卡瓦椒(Kava)、半边莲(Lobelia)和育亨宾(Yohimbe)。 据报告,这12种保健食品虽不是最畅销的,但却是市面上相当普遍的,在全美各地一般健康食品专卖店都能买到。《消费者报道》指出,常常被用来消炎、治疗伤口与关节疼痛的乌头草,对于人体健康的潜在威胁是可能具有毒性,会导致恶心、呕吐、低血压、呼吸系统麻痹、心律不齐甚至死亡;具有减肥、治疗鼻塞等效果的苦橙,副作用是可能导致昏厥、心律不齐、中风甚至死亡;榭树常被用来治疗感冒,且有排毒效果,但副作用是可能损害肝脏、肾脏;胶体银有助减轻细菌感染或食物中毒,却可能让人皮肤变成蓝色、黏膜变色以及损害肾脏;款冬可减轻咳嗽、喉咙痛、气喘,潜在威胁是可能留下肝脏问题或导致癌症;可治咳嗽的紫草,不良作用是造成肝脏问题或导致癌症;紫罗兰对鼻塞、支气管炎等有舒缓效果,却可能引发心脏病、中风、心律不齐甚至死亡;常被用来减轻疼痛、消炎的锗,不但可能损害肾脏,也可能致死;可以减轻肠胃不适的白屈菜,潜在威胁是可能带来肝脏损坏;被认为有降低焦虑效果的卡瓦椒,潜在威胁是可能损坏肝脏;半边莲能消除咳嗽,但具有毒性,服用过量会导致心跳加速、血压降到极低、陷入昏迷甚至可能致死;育亨宾被认为可改善性器官勃起障碍,并可减轻胸部疼痛,服用一般剂量可能造成血压高、心跳加速,如果服用过量则可能导致血压严重降低,产生心脏问题,甚至死亡。
乌头碱标准品配制好后放在冰箱里过了2周左右就分解了,就不能用于薄层了。我用的溶剂是无水乙醇1mg配制成2ml。请问各位要怎么贮存才不容易分解。
日前,美国权威消费杂志《消费者报道》(Consumer Reports)发布的调查报告指出,由于缺乏具体规范,有些产品甚至含重金属、杀虫剂与处方类药品。与此同时消费者却不知道贩售这些健康食品的业者其实并不能证实这些产品确实安全有效。《消费者报道》指出,包括乌头草在内的12种草药制成的保健食品可能对人体造成不良影响,消费者应避免服用。究竟是因为这些原料不符合规范导致重金属,杀虫剂超标,还是因为这些原料本身的某种成分对人体可能造成不良影响?如果这些不能分清楚,从而就简单的将这些归于对人体有害一列,对这些原料而言,有失公平。个人观点认为说的更像是“是药三分毒”的这个道理,把药当饭吃,肯定会有副作用的。国内的此类产品是否有同样的问题?重金属,杀虫剂等超标?不知有哪些标准规范适用于这些原材料。欢迎大家提供相关标准规范,发表自己的意见。 这12种保健食品原料包括:乌头草(Aconite)苦橙(BitterOrange)榭树(Chaparral)胶体银(ColloidalSilver)款冬(Coltsfoot)紫草(Comfrey)紫罗兰(CountryMallow)锗(Germanium)白屈菜(GreaterCelandine)卡瓦椒(Kava)半边莲(Lobelia)育亨宾(Yohimbe)。
现在应该说这一点是清楚的:氰戊菊酯其中一个峰一定会与顺式氰戊菊酯重合,无论采取什么分离方式。因为氰戊菊酯本身就包含顺式氰戊菊酯。这种说法应该没错吧?问题是,我现在就是需要计算同一个茶样中,氰戊菊酯和顺式氰戊菊酯2个项目的含量。氰戊菊酯的含量我一直有做,就是用氰戊菊酯的标曲来计算,每个浓度的氰戊菊酯标样都有2个峰,计算时用的是组校准,而非浓度合计。而我的疑问就是:不论是出几个峰,茶样中顺式氰戊菊酯的含量该如何计算?应该用顺式氰戊菊酯的标样作曲线,从而计算含量;而不应该是直接用氰戊菊酯的标曲计算,对吧?不好意思,脑袋愚笨,多有打扰!!!
做项目氰戊菊酯和顺式氰戊菊酯时,进标准品是只进氰戊菊酯还是单独两个都进?(氰戊菊酯后面那个峰不就是顺式氰戊菊酯吗?)
顺式氰戊菊酯进GC-uECD应该出几个峰?我进了针顺式氰戊菊酯单标(标准物质中心购买),居然出来了两个峰,同时在下一针还出现了一个鬼峰~安捷伦7890A DB17-30M。图见附件。
[color=#444444]现在用甲醇水缓冲盐,各种比例试过了,无法分离,甚至就是重叠的。混标根本没有分开的意思。[/color][color=#444444][color=#444444]现在就用液相色谱,没有质谱,甲醇和磷酸二氢钾缓冲液比例从小到大,其他物质都分开了,尿刊酸的顺式和反式的同一保留时间,混标和单标一致[/color][/color]
hp-5柱子,方法sn/t1117-2008,跑出来氰戊菊酯和顺式氰戊菊酯都是三个峰,标准上是两个,老师来帮助解答下疑惑。
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font][font=宋体] 取本品粉末2g,加氨试液2ml润湿,加乙醚20ml,超声处理30分钟,滤过,滤液挥干,残渣加二氯甲烷1ml使溶解,作为供试品溶液。另取苯甲酰乌头原碱对照品、苯甲酰次乌头原碱对照品、[color=var(--weui-LINK)]苯甲酰新乌头原碱[i][/i][/color]对照品,加异丙醇-三氯甲烷(1:1)混合溶液制成每1ml各含1mg的[color=var(--weui-LINK)]混合溶液[i][/i][/color],作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5[/font]μ[font=宋体]l[/font][font=宋体],分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯-甲醇(6.4:3.6:1)为展开剂,置氨蒸气饱和20分钟的展开缸内,展开,取出,晾干,喷以稀碘化铋钾试液。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[/font][font=宋体][/font][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font] [b][font=宋体][/font][font=宋体][/font] [font=宋体]水分[/font][/b][font=宋体] [/font][font=宋体]不得过12.0%(通则0832第二法)。[/font] [b][font=宋体]双酯型生物碱[/font][/b][font=宋体] [/font][font=宋体]照〔含量测定〕项下色谱条件和供试品溶液的制备方法试验。[/font] [b][font=宋体]对照品溶液的制备 [/font][/b][font=宋体]取[color=var(--weui-LINK)]乌头碱[i][/i][/color]对照品、次乌头碱对照品及新乌头碱对照品适量,精密称定,加异丙醇-三氯甲烷(1:1)混合溶液分别制成每1ml含乌头碱30[/font]μ[font=宋体]g[/font][font=宋体]、次乌头碱10[/font]μ[font=宋体]g[/font][font=宋体]、新乌头碱50[/font]μ[font=宋体]g[/font][font=宋体]的溶液,即得。[/font] [b][font=宋体]测定法 [/font][/b][font=宋体]分别精密吸取对照品溶液与〔含量测定〕项下供试品溶液各10[/font]μ[font=宋体]l[/font][font=宋体],注入[color=var(--weui-LINK)][url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url][i][/i][/color]仪,测定,即得。[/font] [font=宋体]本品含双酯型生物碱以乌头碱([/font]C[sub]34[/sub]H[sub]47[/sub]NO[sub]11[/sub][font=宋体])、次乌头碱([/font]C[sub]33[/sub]H[sub]45[/sub]NO[sub]10[/sub][font=宋体])和新乌头碱([/font]C[sub]33[/sub]H[sub]45[/sub]NO[sub]11[/sub][font=宋体])的总量计,不得过0.040%。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以十八烷基硅烷键合硅胶为填充剂;以乙腈-四氢呋喃(25:15)为流动相A,以0.1mol/L醋酸铵溶液(每1000ml加冰醋酸0.5ml)为流动相B,按下表中的规定进行梯度洗脱;检测波长为235nm。理论板数按苯甲酰新乌头原碱峰计算应不低于2000。[/font] [align=center] [/align] [b][font=宋体]对照品溶液的制备 [/font][/b][font=宋体] [/font][font=宋体]取苯甲酰乌头原碱对照品、苯甲酰次乌头原碱对照品、苯甲酰新乌头原碱对照品适量,精密称定,加异丙醇-三氯甲烷(1:1)混合溶液分别制成每1ml含苯甲酰乌头原碱20[/font]μ[font=宋体]g[/font][font=宋体]、苯甲酰次乌头原碱0.1mg、苯甲酰新乌头原碱80[/font]μ[font=宋体]g[/font][font=宋体]的混合溶液,即得。[/font] [b][font=宋体]供试品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取本品粉末(过三号筛)约2g,精密称定,置具塞锥形瓶中,加氨试液3ml,精密加入异丙醇-乙酸乙酯(1:1)混合溶液50ml,称定重量,超声处理(功率300W,频率40kHz;水温在25℃以下)30分钟,放冷,再称定重量,用异丙醇-乙酸乙酯(1:1)混合溶液补足减失的重量,摇匀,滤过。精密量取续滤液25ml,40℃以下减压回收溶剂至干,残渣精密加入异丙醇-三氯甲烷(1:1)混合溶液3ml溶解,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]分别精密吸取对照品溶液与供试品溶液各10[/font]μ[font=宋体]l[/font][font=宋体],注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,即得。[/font] [font=宋体]本品按干燥品计算,含苯甲酰乌头原碱([/font]C[sub]32[/sub]H[sub]45[/sub]NO[sub]10[/sub][font=宋体])、苯甲酰次乌头原碱([/font]C[sub]31[/sub]H[sub]43[/sub]NO[sub]9[/sub][font=宋体])及苯甲酰新乌头原碱([/font]C[sub]31[/sub]H[sub]43[/sub]NO[sub]10[/sub][font=宋体])的总量应为0.020%~0.070%。[/font] [font=宋体] [/font]
大家好,下面是我的液相条件,其中乌头碱和次乌头碱没有分开,另外所有峰均拖尾,我该怎么调整?谢谢各位了条件 流速:1ml/min 柱温:室温 时间 乙腈 5mmol/L乙酸铵 (25微升冰醋酸)0min 2%15 min 19%15.1 min 22%30 min 35%55 min 35 % 保留时间苯甲酸 9.8min苯甲酰新乌头原碱 26.1 min苯甲酰乌头原碱 28.7 min苯甲酰次乌头原碱 30.2 min新乌头碱 36.4 min乌头碱 43 min次乌头碱 42.8 min
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][font=宋体](1)本品横切面:后生皮层为7~8列棕黄色栓化细胞;皮层有[color=var(--weui-LINK)]石细胞[i][/i][/color],单个散在或2~5个成群,类长方形、方形或长圆形,胞腔大;内皮层明显。韧皮部宽广,常有不规则裂隙,筛管群随处可见。形成层环呈不规则多角形或类圆形。木质部导管1~4列或数个相聚,位于形成层角隅的内侧,有的内含棕黄色物。髓部较大。薄壁细胞充满淀粉粒。[/font][font=宋体]粉末灰棕色。淀粉粒单粒类圆形,直径2~23[/font][font=&]μ[/font][font=宋体]m[/font][font=宋体];复粒由2~16分粒组成。石细胞无色,与后生皮层细胞连结的显棕色,呈类方形、类长方形、类圆形、梭形或长条形,直径20~133(234)[/font][font=&] μ[/font][font=宋体]m[/font][font=宋体],长至465[/font][font=&]μ[/font][font=宋体]m[/font][font=宋体],壁厚薄不一,壁厚者层纹明显,纹孔细,有的含棕色物。后生皮层细胞棕色,表面观呈类方形或长多角形,壁不均匀增厚,有的呈瘤状突入细胞腔。[/font][font=宋体](2)取本品粉末1g,加氨试液2ml润湿,加乙醚20ml,超声处理30分钟,滤过,滤液挥干,残渣加异丙醇-三氯甲烷(1:1)混合溶液1ml使溶解,作为供试品溶液。另取乌头双酯型生物碱对照提取物,加异丙醇-三氯甲烷(1:1)混合溶液制成每1ml各含3mg的混合溶液,作为对照提取物溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液5μl,对照提取物溶液10μl,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯-甲醇(6.4:3.6:1)为展开剂,置氨蒸气预饱和20分钟的展开缸内,展开,取出,晾干,喷以稀碘化铋钾试液,在日光下检视。供试品色谱中,在与对照提取物色谱相应的位置上,显相同颜色的斑点。[/font][size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][b][font=宋体]杂质[/font][/b][font=宋体](残茎)[/font][font=&] [/font][font=宋体] [/font][font=宋体]不得过5%(通则2301)。[/font][b][font=宋体]水分 [/font][/b][font=&] [/font][font=宋体]不得过12.0%(通则0832第二法)。[/font][b][font=宋体]总灰分 [/font][/b][font=&] [/font][font=宋体]不得过6.0%(通则2302)。[/font][b][font=宋体]【含量测定】[/font][/b][font=宋体] 照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font][b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以[color=var(--weui-LINK)]十八烷基硅烷键合硅胶[i][/i][/color]为填充剂;以乙腈为流动相A,以0.2%冰醋酸溶液(三乙胺调节pH值至6.20)为流动相B,按下表中的规定进行梯度洗脱;检测波长为235nm。理论板数按新乌头碱峰计算应不低于2000。[/font][b][font=宋体]对照提取物溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取乌头双酯型生物碱对照提取物(已标示新乌头碱、次乌头碱和乌头碱的含量)20mg,精密称定,置10ml量瓶中,加0.01%盐酸乙醇溶液使溶解并稀释至刻度,摇匀,即得。[/font][b][font=宋体]标准曲线的制备[/font][/b][font=宋体] [/font][font=宋体]精密量取上述对照提取物溶液各1ml,分别置2ml,5ml,10ml,25ml量瓶中,加0.01%盐酸乙醇溶液稀释至刻度,摇匀。分别精密量取对照提取物溶液及上述系列浓度对照提取物溶液各10μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,以对照提取物中相当于新乌头碱、次乌头碱和乌头碱的浓度为横坐标,相应色谱峰的峰面积值为纵坐标,绘制标准曲线。[/font][b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]取本品粉末(过三号筛)约2g,精密称定,置具塞锥形瓶中,加氨试液3ml,精密加入异丙醇-乙酸乙酯(1:1)混合溶液50ml,称定重量,超声处理(功率300W,频率40kHz;水温25℃以下)30分钟,放冷,再称定重量,用异丙醇-乙酸乙酯(1:1)混合溶液补足减失的重量,摇匀,滤过。精密量取续滤液25ml,40℃以下减压回收溶剂至干,残渣加0.01%盐酸乙醇溶液使溶解,转移至5ml量瓶中,并稀释至刻度,摇匀,滤过,精密吸取10μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,按标准曲线计算,即得。[/font][font=宋体]本品按干燥品计算,含乌头碱(C[sub]34[/sub]H[sub]47[/sub]NO[sub]11[/sub])、次乌头碱(C[sub]33[/sub]H[sub]45[/sub]N0[sub]10[/sub])和新乌头碱(C[sub]33[/sub]H[sub]45[/sub]NO[sub]11[/sub])的总量应为0.15%~0.75%。[/font][font=宋体][/font]
我用的是岛津GC2010-ECD,RTX-50的色谱柱。无论如何改变条件(减低温度和升温速率),氰戊菊酯和顺式氰戊菊酯就是重叠在一起,分不开。不知大虾们有何方法可以将其分离?谢谢!
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font][/b][font=宋体][/font][b][font=宋体][/font][/b] [font=宋体] [/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体](1)本品横切面:后生皮层为7~8列棕黄色栓化细胞;皮层有[color=var(--weui-LINK)]石细胞[i][/i][/color],单个散在或2~5个成群,类长方形、方形或长圆形,胞腔大;[color=var(--weui-LINK)]内皮层[i][/i][/color]明显。韧皮部宽广,常有不规则裂隙,筛管群随处可见。形成层环呈不规则多角形或类圆形。木质部导管1~4列或数个相聚,位于形成层角隅的内侧,有的内含棕黄色物。髓部较大。薄壁细胞充满淀粉粒。[/font] [font=宋体]粉末灰棕色。淀粉粒单粒类圆形,直径2~23[/font][font=&]μ[/font][font=宋体]m[/font][font=宋体];复粒由2~16分粒组成。石细胞无色,与后生皮层细胞连结的显棕色,呈类方形、类长方形、类圆形、梭形或长条形,直径20~133(234)[/font][font=&] μ[/font][font=宋体]m[/font][font=宋体],长至465[/font][font=&]μ[/font][font=宋体]m[/font][font=宋体],壁厚薄不一,壁厚者层纹明显,纹孔细,有的含棕色物。后生皮层细胞棕色,表面观呈类方形或长多角形,壁不均匀增厚,有的呈瘤状突入细胞腔。[/font] [font=宋体](2)取本品粉末1g,加氨试液2ml润湿,加乙醚20ml,超声处理30分钟,滤过,滤液挥干,残渣加异丙醇-三氯甲烷(1:1)混合溶液1ml使溶解,作为供试品溶液。另取乌头双酯型生物碱对照提取物,加异丙醇-三氯甲烷(1:1)混合溶液制成每1ml各含3mg的混合溶液,作为对照提取物溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液5μl,对照提取物溶液10μl,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯-甲醇(6.4:3.6:1)为展开剂,置氨蒸气预饱和20分钟的展开缸内,展开,取出,晾干,喷以稀碘化铋钾试液,在日光下检视。供试品色谱中,在与对照提取物色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][b][font=宋体][/font][/b] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体]杂质[/font][/b][font=宋体](残茎)[/font][font=&] [/font][font=宋体] [/font][font=宋体]不得过5%(通则2301)。[/font] [b][font=宋体]水分 [/font][/b][font=&] [/font][font=宋体]不得过12.0%(通则0832第二法)。[/font] [b][font=宋体]总灰分 [/font][/b][font=&] [/font][font=宋体]不得过6.0%(通则2302)。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以[color=var(--weui-LINK)]十八烷基硅烷键合硅胶[i][/i][/color]为填充剂;以乙腈为流动相A,以0.2%冰醋酸溶液(三乙胺调节pH值至6.20)为流动相B,按下表中的规定进行梯度洗脱;检测波长为235nm。理论板数按新乌头碱峰计算应不低于2000。[/font] [align=center] [/align] [b][font=宋体]对照提取物溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取乌头双酯型生物碱对照提取物(已标示新乌头碱、次乌头碱和乌头碱的含量)20mg,精密称定,置10ml量瓶中,加0.01%盐酸乙醇溶液使溶解并稀释至刻度,摇匀,即得。[/font] [b][font=宋体]标准曲线的制备[/font][/b][font=宋体] [/font][font=宋体]精密量取上述对照提取物溶液各1ml,分别置2ml,5ml,10ml,25ml量瓶中,加0.01%盐酸乙醇溶液稀释至刻度,摇匀。分别精密量取对照提取物溶液及上述系列浓度对照提取物溶液各10μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,以对照提取物中相当于新乌头碱、次乌头碱和乌头碱的浓度为横坐标,相应色谱峰的峰面积值为纵坐标,绘制标准曲线。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]取本品粉末(过三号筛)约2g,精密称定,置具塞锥形瓶中,加氨试液3ml,精密加入异丙醇-乙酸乙酯(1:1)混合溶液50ml,称定重量,超声处理(功率300W,频率40kHz;水温25℃以下)30分钟,放冷,再称定重量,用异丙醇-乙酸乙酯(1:1)混合溶液补足减失的重量,摇匀,滤过。精密量取续滤液25ml,40℃以下减压回收溶剂至干,残渣加0.01%盐酸乙醇溶液使溶解,转移至5ml量瓶中,并稀释至刻度,摇匀,滤过,精密吸取10μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,按标准曲线计算,即得。[/font] [font=宋体]本品按干燥品计算,含乌头碱(C[sub]34[/sub]H[sub]47[/sub]NO[sub]11[/sub])、次乌头碱(C[sub]33[/sub]H[sub]45[/sub]N0[sub]10[/sub])和新乌头碱(C[sub]33[/sub]H[sub]45[/sub]NO[sub]11[/sub])的总量应为0.15%~0.75%。[/font]
最近用的方法检测菊酯类农药氟氰戊菊酯、溴氰菊酯及顺式氰戊菊酯回收率超低的,请问有什么好的方法做比较好
请问顺式氰戊菊酯和反式氰戊菊酯谁先出峰?好友回复:哪个极性大呀大家说说~
燃烧头和雾化器清洗的时候能用稀酸浸泡吗?燃烧头能用超声波清洗吗?
川乌,正名为乌头,别名:鹅儿花、铁花、五毒;被子植物门,毛茛科,乌头属。英文名Aconite Root, 生于山地草坡或灌丛中。栽培于平地肥沃的砂质土壤,主要栽培于四川。分布于长江中下游,北至秦岭和山东东部,南至广西北部。我们今天给大家带来的应用就是川乌中新乌头碱、次乌头碱、乌头碱等相关成分的测定。[b]色谱条件色谱柱:月旭Ultimate ODS-3(4.6×250mm,5μm)流动相: [/b][table=511][tr][td=1,1,165]时间(min)[/td][td=1,1,136]A(%)[/td][td=1,1,210]B(%)[/td][/tr][tr][td=1,1,165]0[/td][td=1,1,136]21[/td][td=1,1,210]79[/td][/tr][tr][td=1,1,165]44[/td][td=1,1,136]31[/td][td=1,1,210]69[/td][/tr][tr][td=1,1,165]65[/td][td=1,1,136]35[/td][td=1,1,210]65[/td][/tr][tr][td=1,1,165]70[/td][td=1,1,136]35[/td][td=1,1,210]65[/td][/tr][/table]检测波长:235nm柱温:30℃流速:1.0mL/min进样量:10μL[b]谱图和数据[/b][color=#333333]1、对照溶液图谱[/color][align=center][b][color=#333333][img=,600,325]https://ng1.17img.cn/bbsfiles/images/2019/09/201909160929442473_6839_932_3.jpg!w690x374.jpg[/img][img=,600,93]https://ng1.17img.cn/bbsfiles/images/2019/09/201909160929509953_4253_932_3.jpg!w690x108.jpg[/img][/color][/b][/align][align=center][b][color=#333333][b]结 论[/b][/color][/b][/align][b][color=#333333][b][color=#3e3e3e]使用月旭Ultimate ODS-3(4.6×250mm,5μm),在此色谱条件下,能满足检测需求。[/color][/b][/color][/b]
很多种类的极性化合物分离条件。 UDP-葡萄糖 UDP-葡萄糖、UDP-半乳糖、磷酸半乳糖 葡萄糖 蔗糖 红细胞中的UDP-葡萄糖、UDP-半乳糖、三磷酸腺苷(ATP) ADP-葡萄糖、CDP-葡萄糖 糖核苷酸 胞嘧啶、胸腺嘧啶、尿嘧啶、鸟嘌呤、腺嘌呤 三磷酸腺苷(ATP)、一磷酸腺苷(AMP) 黄嘌呤-磷酸、鸟嘌呤-三磷酸 体液中的黄嘌呤、尿酸、次黄嘌呤 色胺、五羟色胺、多巴胺 L-天冬氨酸、L-精氨酸 L-精氨酸、L-赖氨酸、L-组氨酸 谷氨酸、赖氨酸 亮氨酸、异亮氨酸 L-甲硫氨酸、L-谷氨酸 甲基琥珀酸、戊二酸、草酸、肌酸、4-羟脯氨酸、天冬氨酸、鸟氨酸 叶酸 抗坏血酸 胆汁酸 柠檬酸、马来酸、反式乌头酸 马来酸、富马酸 3-羟基肉桂酸 矮壮素、甲哌啶 苯海拉明 4-二甲氨基吡啶 草甘膦 三聚氰胺、三聚氰酸 胍
很多种类的极性化合物分离条件。 UDP-葡萄糖 UDP-葡萄糖、UDP-半乳糖、磷酸半乳糖 葡萄糖 蔗糖 红细胞中的UDP-葡萄糖、UDP-半乳糖、三磷酸腺苷(ATP) ADP-葡萄糖、CDP-葡萄糖 糖核苷酸 胞嘧啶、胸腺嘧啶、尿嘧啶、鸟嘌呤、腺嘌呤 三磷酸腺苷(ATP)、一磷酸腺苷(AMP) 黄嘌呤-磷酸、鸟嘌呤-三磷酸 体液中的黄嘌呤、尿酸、次黄嘌呤 色胺、五羟色胺、多巴胺 L-天冬氨酸、L-精氨酸 L-精氨酸、L-赖氨酸、L-组氨酸 谷氨酸、赖氨酸 亮氨酸、异亮氨酸 L-甲硫氨酸、L-谷氨酸 甲基琥珀酸、戊二酸、草酸、肌酸、4-羟脯氨酸、天冬氨酸、鸟氨酸 叶酸 抗坏血酸 胆汁酸 柠檬酸、马来酸、反式乌头酸 马来酸、富马酸 3-羟基肉桂酸 矮壮素、甲哌啶 苯海拉明 4-二甲氨基吡啶 草甘膦 三聚氰胺、三聚氰酸 胍
云南省大理州政府食品安全政务微博发布消息,据宾川县人民政府新闻办公室通报,9月8日晚,宾川县金牛镇一村民邀约亲戚朋友到家中煮食草乌炖猪脚,参加就餐的亲属先后出现中毒症状,并到医院就诊,经医院全力抢救,其中6人抢救无效死亡,其余21人正在救治当中。 接到“有村民食用草乌中毒”的报告后,宾川县立即启动公共突发事件应急预案,一方面全力开展医疗救治工作,另一方面对当天参加就餐的人员进行全面排查,对出现疑似中毒症状的人员及时送医院救治。 同时,再次在全县范围内发出通知,要杜绝食用不明食材,防止类似中毒事件的发生。 目前,宾川县正全力开展救治工作。有这么吃的么?生草乌含有乌头碱、新乌头碱、次乌头碱一类的生物碱成分。这类成分具有很大的毒性,口服乌头碱几毫克即可致命。中毒时可出现麻痹、心率失常、低血压、呼吸困难等表现。
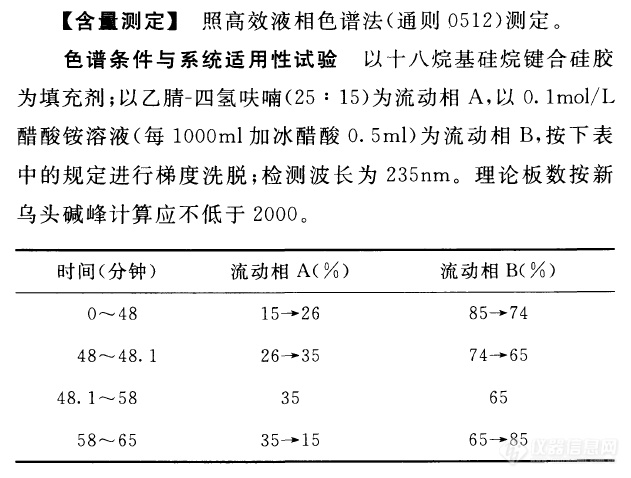
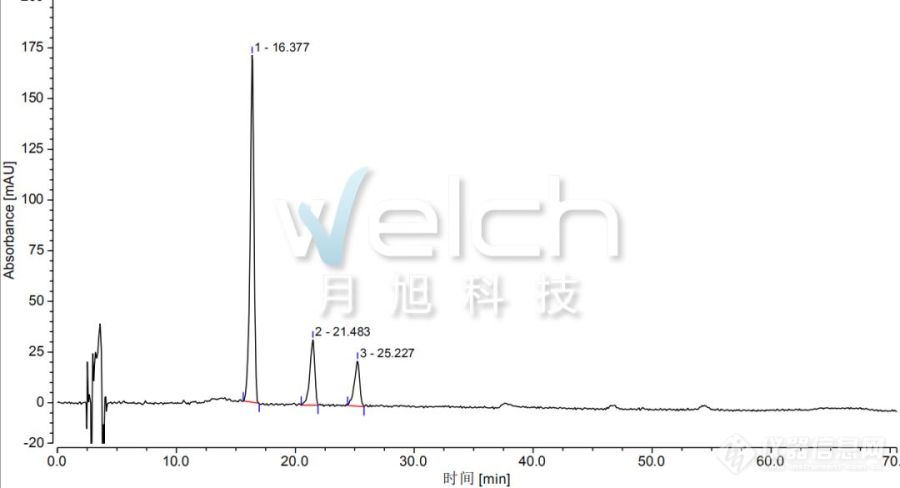
笔者以前刚进入仪器行业时的工作是[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]应用工程师。有次去客户那做样,是分析草乌中的乌头碱、次乌头碱及新乌头碱,当时参考的还是2015版药典。在2015版药典中,乌头碱、次乌头碱与新乌头碱的测试方法如下:色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-四氢呋喃(25:15)为流动相A,以0.1mol/L醋酸铵溶液(每1000ml加冰醋酸0.5ml)为流动相B,按下表中的规定进行梯度洗脱;检测波长为235nm。理论板数按新乌头碱峰计算应不低于2000。 ----------------------------------------- 时间(分钟) 流动相A(%) 流动相B(%) ----------------------------------------- 0~48 15→26 85→74 48~48.1 26→35 74→65 48.1~58 35 65 58~65 35→15 65→85-----------------------------------------[align=center][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211301550528624_1221_1809927_3.png[/img][/align]当时使用的色谱柱是日本的一款色谱柱加相应的保护柱,柱效超高,新乌头碱的理论板数过万。就是在此时测试中,开始时压力为10Mpa,在做了四五个样品后,压力超过了20Mpa,很明显是发生了堵塞现象,于是进行排查,从流动相开始沿着流路逐级排查。最后发现是保护柱出了问题,很多黄白色的泥状物堵塞在保护柱的入口筛板处,产生了堆积,最后导致压力越来越高。这个问题只在草乌、川乌等检测乌头碱、次乌头碱与新乌头碱时才会出现,其他应用时并未出现。于是对泥状物进行清理,发现压力恢复到最初的10Mpa,但是多针之后又出现此现象。在目前的2020版药典中,草乌的测试条件相对于2015版药典有了较大的变化,我们可以看出,新版去掉了四氢呋喃,流动相B也发生了较大变化,相对于2015版的方法,可以认为是个全新的方法了。不知道对于我原本使用过的那款保护柱,使用现在的方法测试草乌中的生物碱还堵不堵了。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211301550531227_7420_1809927_3.png[/img][/align]
最近正做顺式氯氰菊酯的标准曲线,可是我用0.01ppm的标样进针却不出峰。只有10ppm的才出峰,而且这个峰出完。在进一针溶剂时还会又峰出现,要用溶剂冲洗号几次才没有峰出现。 请问这是什么原因?各位知道的或是又过同样经历的指点一下 谢谢[font=楷体_GB2312]楼主的仪器的条件是:4890d DB-1的柱子 柱头压是50kpa;总流速:60ml/min;隔垫吹扫流速:3ml/min;进样口:260;检测器:300;柱温:60保持1min 以15℃/min到220℃ 保持5min 以15℃/min到280℃保持10min 分析顺式氯氰菊酯 做标准曲线时0.01ppm的不出峰 只有0.5ppm的出峰。但是我的药的MRL值是0.05ppm [/font]
我的样品就是乌头碱类化合物,现在证明四氢呋喃起到了改善峰型作用,但是醋酸铵又有问题了。2010版药典中对乌头碱类的流动相体系是乙腈-四氢呋喃-醋酸铵-冰醋酸体系。其中醋酸铵是0.1mol/L(每1000ml中含冰醋酸0.5ml),现在的问题是醋酸铵的浓度过大,在运行过程中会析出堵了阀中的滤芯,柱压一直升高,我该怎么办呢?我试过将醋酸铵的浓度减到0.03mol/L,但是峰型又会不好,会有峰前沿出现。该如何改善呢?感谢各位了!







 我要推广仪器
我要推广仪器
 下载APP
下载APP