[color=#444444]网上查询邻溴苯硫酚沸点:194~196摄氏度,对溴苯硫酚沸点:239摄氏度,色谱柱为Rtx_624,设进样口温度250,检测器270,柱溫160~230的范围,也走过程序升温,分离度最好才是1.0,不知道怎么实验怎么做下去了,请指导一二,另外,准备购买一根极性柱子,思路对不?[/color]
[color=#444444]2,4-二甲基苯硫酚,2,6-二甲基苯硫酚位置异构体。沸点分别为207-208 °C,76℃[/color][color=#444444]使用Scientific TRACE TR-5MS 色谱柱,两个峰出在一起,相差0.1min,无法基线分离。尝试多种升温程序,只有保留时间和峰宽有变化,分离情况不变。降低载气流速也一样。[/color][color=#444444]换用 安捷伦vf-WAXms柱,彻底分不开了。瘦高的一个峰。[/color][color=#444444]两个柱子都试过60,70,90,120度恒温,分不开。梯度升温,也分不开。[/color][color=#444444]请各位大侠帮帮忙,试了两天了。[/color]
邻氨基苯硫酚能不能走液相色谱?有什么要求吗?
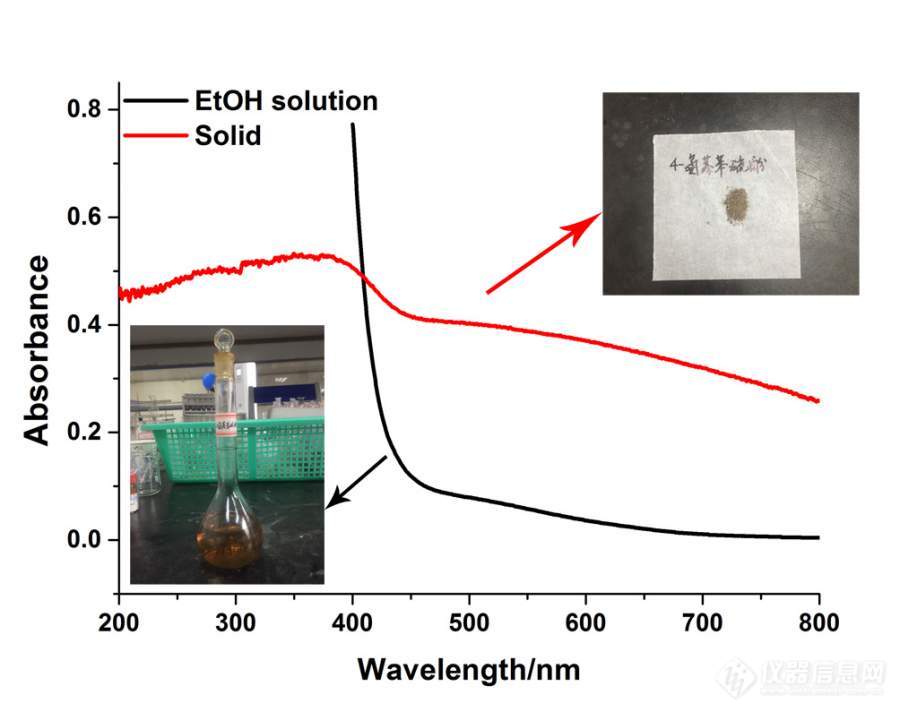
[color=#444444]我最近在做关于对氨基苯硫酚(4-ATP)的实验,我在实验中发现,这个本身是棕褐色的,无论在乙醇溶液还是固体状态,但是它在紫外检测条件下的光谱我发现有些不同。固体条件下,它在可见光区是有着吸收的,但是在乙醇溶液中,可见光的范围内,它的吸收非常低。图中的溶液浓度是10exp(-2)M。我想问问各位,这个应该怎么解释?[/color][color=#444444][/color][color=#444444][img=,690,545]https://ng1.17img.cn/bbsfiles/images/2019/09/201909111034332125_8799_1801607_3.jpg!w690x545.jpg[/img][/color][color=#444444][color=#008000]对氨基苯硫酚(4-ATP).jpg[/color][/color]
卤代酚卤代酚是含酚的卤代化合物,对革兰氏阳性菌有强杀菌作用,用在化妆品中的卤代酚有六氯酚 等多种化合物。这类化合物通常是光敏物质。我国化妆品卫生标准规定为限用物质,限用量见表2-3-17。表 2-3-17 化妆品卫生标准中卤代酚的限用量品名 序号 最大使用量(%) 溴氯双酚 4-4 0.1 双氯酚 4-7 0.2 2,4-二氯二甲苯酚 4-8 0.1 三氯生 4-21 0.3 六氯酚 4-24 0.1 4-溴邻甲苯酚 4-31 0.3 苄氯酚 4-42 0.2 4-氯2-甲苯酚 4-55 0.2 4-氯3,5-二甲苯酚 4-56 0.2 * 指化妆品卫生标准(GB7916-87)中的序号,4-42即表4的序号42(一)薄层色谱法(TLC)1 适用范围本方法适用于化妆品中六氯酚,双二氯酚硫醚、二氯酚和三溴水杨酞替苯胺的定性。2 原理样品经预处理后,样液中的卤代酚用的薄层色谱法进行分离、呈色,然后与标准斑点比较,进行定性。3 试剂3.1 乙醇:分析纯。3.2 己烷:分析纯。 3.3 丙酮:分析纯。3.4 无水硫酸钠:分析纯。3. 5硫酸(lmol/L)。3.6 六氯酚标准溶液(1):准确称取用苯重结晶的六氯酚50.0mg,加丙酮溶解后移入50ml容量瓶中并定容至刻度,避光保存。此溶液1ml含1.0mg六氯酚。3.7 双二氯酚硫醚(2):准确称取用苯重结晶的双二氯酚硫醚50.omg,用丙酮溶解,移入50ml容量瓶中并定容至刻度,避光保存。此溶液lml含1.0mg二氯酚硫醚。3.8双氯酚标准溶液(3):准确称取用甲苯重结晶的双氯酚50.0mg,用丙酮溶解,移入50ml容量瓶中,定容至刻度。此溶液1.0ml含1.0mg二氯酚,避光保存。3.9三溴水杨酞替苯胺(4):准确称取用丙酮重结晶的三溴水杨酞替苯胺50.0mg,用丙酮溶解,移入50ml容量瓶中并定容至刻度。此溶液1.0ml含1.0mg三溴水杨酞替苯胺,避光保存。3.10 乙醇一己烷(1 9)。3.ll离子交换纤维素(5):将DEAE(二乙基氨基乙醇)纤维素,(交换量约0.9meg/g),浸泡于50倍量的0.lmol/L的盐酸中,用玻璃漏斗过滤,用20倍量的丙酮,30倍量的0.lmo1/L氢氧化钠溶液淋洗至OH-型后,用水洗成中性,再用20倍量的丙酮淋洗,弃去丙酮。空气中干燥。保存在乙醇十己烷(l+9)溶液中。3.12 硅胶:薄层用硅胶中加有荧光剂。3.13碱性氧化铝。3.14展开剂:石油醚 冰乙酸(89 12)3.15显色剂。3.15.1 浓氨水。3.15.2 2%4-氨基安替比林溶液:称取2g4-氨基安替比林用乙醇溶解稀释至100ml。3.15.3 8%铁氰化钾溶液(K3[Fe(CN)6])。3.15.4 2%三氯化铁溶液(FeCl36H20):称取2g三氯化铁用乙醇溶解稀释至100ml。3.15.5 2%铁氰化钾溶液。3.16 盐酸 丙酮溶液:9.5ml盐酸加丙酮至100ml(临用前配制)。4 仪器 4.1 层析柱:、内径10mm、高200mm的具塞玻璃管的下端熔接玻璃过滤器或塞有玻璃棉,4.2 紫外灯,具有8W功率,254nm波长。4.3离子交换柱(6):将离子交换纤维素用乙醇 已烷(3.11)配成混悬液,:用湿式填充法缓慢倾入层析柱中,以防止产生气泡,填充高度80mm。5 分析步骤5.1样品预处理(7)(8)称取含卤化酚0.5mg的样品(扑粉,除臭砂芯、香波约10g,膏霜约0.5g),置于100ml玻璃瓶中,连接好回流冷凝器:加50ml乙醇 已烷(1 9)溶液,2ml 1mol/L硫酸,于水浴上加热3min,冷却后用3号玻璃砂芯漏斗过滤,用乙醇 己烷(1 9)溶液5ml洗沉淀,滤液移入分液漏斗中静置分层。取己烷层用10ml水洗涤,无水硫酸钠脱水后以0.5ml/min的流速注入离子交换柱(10)。用50ml己烷洗涤。去除油脂等干扰物质,弃去淋洗液,依次用10ml丙酮、2ml丙酮 盐酸溶液(3.16),20ml丙酮洗脱(11)。溶出液在水溶上加热蒸去有机溶媒,加5ml乙醇,加热使盐酸挥发,重复此操作2次。残渣加2.0ml丙酮溶解,作为样品待测溶液。5.2 制备薄层板5.2.1硅胶薄层板:硅胶30g,加水约65ml,搅拌均匀,涂布成厚度0.25~0.3mm的薄层板,105~l10℃干燥30min,置干燥器中保存。 5.2.2含硝酸银的氧化铝薄层板:0.12g AgNO3,加少量水溶解,加30ml乙醇、20g氧化铝,调成浆状物,涂布厚度为0.25~O.3mm的薄层板,空气中干燥、于干燥器中避光保存。5.3 点样距薄层板底边2cm处将5~20μl待测溶液从左到右点样(12),两点间隔约1cm,薄员板.的右边点2μl标准溶液,空气中干燥。5.4 展开取适量展开剂(3.14)倾人展开槽中,将薄层板放入展开剂中,待溶剂上升约10cm,取出薄层板,空气中干燥。5.5显色(13)在薄层板上顺序喷雾显色剂3.15.1~3.15.3或3.15.4~3.15.5,六氯酚在显色剂3.15.1~3.15.3中为红色,在3.15.4~3.15.5中为蓝色斑点。二氯酚硫醚和三溴水杨酞替苯胺在3.15.1~3.15.5中为紫色,在3.15.4~3.15.5中呈现蓝色斑点。用加荧光剂的硅胶薄层板测定时,各种卤代酚在紫外线照射下,在各自的Rf 值位置上以荧光为背景呈现出暗黑色的斑点。
二硫代氨基甲酸酯/盐的多残留测定方法一直是行业难点,分享一篇苯二硫酚衍生测定二硫代氨基甲酸盐类化合物的新方法,为同行提供有益借鉴。
哪位朋友知道哪里能买到分析纯硫代双乙醇?(250ml装),实验急用!万分感谢!!
哪位朋友哪里有卖硫代双乙醇(硫二甘醇)(分析纯)的,急用,谢谢!!
HJ 1054-2019测二硫代氨基甲酸酯的标准里面只有代森锰锌转换成二硫化碳的系数,请问各位福美双转换成二硫化碳的系数是多少呀?这个系数是怎么得出来的呀?
请教各位大虾,有没有测试四溴双酚A,六溴环十二烷,邻苯二甲酸二异辛酯,邻苯二甲酸二正丁酯的国标或者其他测试方法?小弟比较急,望各位大哥指教。
有哪个实验室在组织实验室间比对求参加!!!如RoHS限制物质,邻苯,玩具总铅,六溴环十二烷,四溴双酚A等

今天在做检测时,看到标准有一个试剂:新蒸馏的苯酚。不知要如何蒸馏?蒸馏的苯酚与未蒸馏的苯酚有什么区别?感谢“四季风”版友提供的图片及资料:把有结晶的苯酚放置100度C的水浴中溶解,然后倒入蒸馏烧瓶中,放入玻璃珠数粒,瓶口放一支带胶塞200度C的水银温度计,塞紧瓶口(见以下草图)。接好冷凝管,放一接收瓶,在电炉上先加热蒸馏至沸点182度C后约2---3分钟左右,拿去第一个接收瓶,另放一接收瓶接收沸点182度C的苯酚蒸馏液(冷却后即为纯苯酚结晶)。http://ng1.17img.cn/bbsfiles/images/2011/09/201109282355_320046_1641058_3.jpg
【序号】:5【作者】:唐磊【题名】:双氢青蒿素—氨基二硫代甲酸酯衍生物的合成【期刊】:宁夏大学【年、卷、期、起止页码】:2017【全文链接】:https://kns.cnki.net/kns8s/defaultresult/index?crossids=YSTT4HG0%2CLSTPFY1C%2CJUP3MUPD%2CMPMFIG1A%2CWQ0UVIAA%2CBLZOG7CK%2CEMRPGLPA%2CPWFIRAGL%2CNLBO1Z6R%2CNN3FJMUV&korder=SU&kw=%E9%9D%92%E8%92%BF%E7%B4%A0%E6%94%B9%E6%80%A7%20%E8%BF%9B%E5%B1%95
铅 双硫腙分光光度法要用到剧毒物氰化钾,市场现在很难买,毒品管理相当严格。而又没钱送样到监测站测试,毕竟是做课题研究,样品太多了。不知这氰化钾能否被其它无剧毒药剂替代呢?
求助各位做过GB7482-87 双硫腙法测水中锌的大佬,有几点问题需要咨询。1、这个方法中要求用无锌水配置各种试剂,想问下用哇哈哈的水能否替代?2、乙酸钠缓冲液和硫代硫酸钠溶液用双硫腙溶液萃取呈绿色,再用CCl4萃取出去多余的双硫腙。这里最后是水相呈无色,有机相呈绿色吗?CCl4的用量是多少?3、试验空白吸光度一般在多少?最好有大佬能私信个标准曲线给个借鉴。知道这个方法相比[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]很麻烦,但领导要求试试,目前只是看了标准想提前问问这些问题。
今天配出来的双硫腙试剂(做铅的,照5009.12,用氯仿配的)发红,稀释后发现颜色很不对(绿中带红)。有几个问题想请教一下:1.将提取液用棉花过滤至500m1分液漏斗中,用1+1盐酸调至酸性--------------------如何调酸性呢?是不是用酚红?用了对双硫腙本身有没有什么影响?2.配置后双硫腙的保护液是什么?3.颜色发红是什么原因?谢谢大家啦!
以K2Cr2O7基准物标定硫代硫酸钠的指示剂是( )。 A、二苯胺 B、次甲基兰 C、KMnO4 D、淀粉溶液
[size=5][b]简介[/b][/size] 苯酚-硫酸法是利用多糖在硫酸的作用下先水解成单糖,并迅速脱水生成糖醛衍生物,然后与苯酚生成橙黄色化合物。再以比色法测定。[size=5][b]原理[/b][/size] 多糖在硫酸的作用下先水解成单糖,并迅速脱水生成糖醛衍生物,然后与苯酚生成橙黄色化合物。再以比色法测定。[size=5][b]试剂[/b][/size] 1. 浓硫酸:分析纯,95.5% 2. 80%苯酚:80克苯酚(分析纯重蒸馏试剂)加20克水使之溶解,可置冰箱中避光长期储存。 3. 6%苯酚:临用前以80%苯酚配制。([b]每次测定均需现配[/b]) 4. 标准葡聚糖(Dextran,瑞典Pharmacia),或分析纯葡萄糖。 5. 15%三氯乙酸(15%TCA):15克TCA加85克水使之溶解,可置冰箱中长期储存。 6. 5%三氯乙酸(5%TCA):25克TCA加475克水使之溶解,可置冰箱中长期储存。 7. 6mol/L 氢氧化钠:120克分析纯氢氧化钠溶于500ml水。 8. 6mol/L 盐酸[size=5][b]操作[/b][/size] 1.制作标准曲线:准确称取标准葡聚糖(或葡萄糖)20mg于500ml容量瓶中,加水至刻度,分别吸取0.4、0.6、0.8、1.0、1.2、1.4、1.6及1.8ml,各以蒸馏水补至2.0ml,然后加入6%苯酚1.0ml及浓硫酸5.0ml,摇匀冷却,室温放置20分钟以后于490nm测光密度,以2.0ml水按同样显色操作为空白,横坐标为多糖微克数,纵坐标为光密度值,得标准曲线。 2.样品含量测定: ①取样品1克(湿样)加1ml 15%TCA溶液研磨,再加少许5%TCA溶液研磨,倒上清液于10毫升离心管中,再加少许5%TCA溶液研磨,倒上清液,重复3次。最后一次将残渣一起到入离心管。[b]注意:总的溶液不要超出10毫升。[/b](既不要超出离心管的容量)。 ②离心,转速3000转/分钟,共三次。第一次15分钟,取上清液。后两次各5分钟取上清液到25毫升锥形比色管中。最后滤液保持18毫升左右。([b]测肝胰腺样品时,每次取上清液时应过滤。因为其脂肪含量大容易夹带残渣。[/b]) ③水浴,在向比色管中加入2毫升6mol/L 盐酸之后摇匀,在96℃水浴锅中水浴2小时。 ④定容取样。水浴后,用流水冷却后加入2毫升6mol/L氢氧化钠摇匀。定容至25毫升的容量瓶中。吸取0.2ml的样品液,以蒸馏补至2.0ml,然后加入6%苯酚1.0ml及浓硫酸5.0ml,摇匀冷却室温放置20分钟以后于490nm测光密度。每次测定取双样对照。以标准曲线计算多糖含量。[size=5][b]注意[/b][/size][b][b](1)此法简单、快速、灵敏、重复性好,对每种糖仅制作一条标准曲线,颜色持久。 (2)制作标准线宜用相应的标准多糖,如用葡萄糖,应以校正系数0.9校正μg数。 (3)对杂多糖,分析结果可根据各单糖的组成比及主要组分单糖的标准曲线的校正系数加以校正计算。 (4)测定时根据光密度值确定取样的量。光密度值最好在0.1——0.3之间。比如:小于0.1之下可以考虑取样品时取2克,仍取0.2ml样品液,如大于0.3可以减半取0.1ml的样品液测定。[/b] [/b]
四溴双酚A和六溴环十二烷有相关ISO EN的标准吗
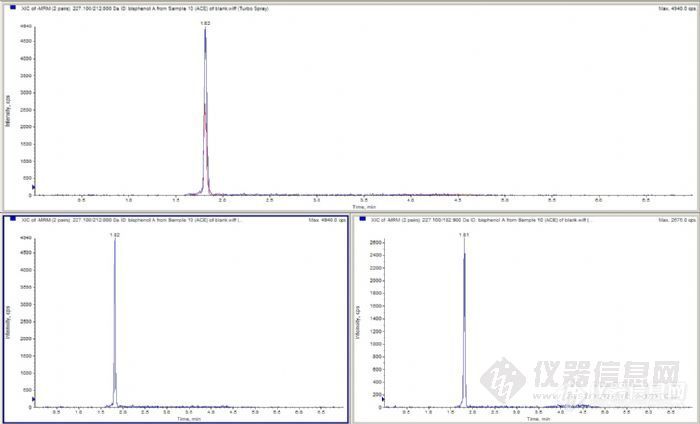
之前做过双酚A等物质的测定,但由于双酚A不在前面项目的研究物质当中,所以虽然同时测定了,但没太注意过双酚A的响应、残留等情况,而且由于存在基质效应,因此生物样本中的双酚A的响应比较低。现在要做水中的双酚A的测定,发现空白水、空白溶剂等直接进样,都会有很高的响应。是不是残留在液相的哪个部分?有没有推荐的溶剂能够清洗一下,或者可以清洗一下仪器的哪个部件?下面是某溶剂的出峰http://ng1.17img.cn/bbsfiles/images/2011/12/201112061403_335920_1644952_3.jpg
硫双威原药该产品有效成分硫双威的结构式和基本物化参数如下:化学名称:3,7,9,13-四甲基-5,11-二氧杂-2,8,14-三噻-4,7,9,12-四-氮杂十五烷-3,12-二烯-6,10-二酮结构式http://ng1.17img.cn/bbsfiles/images/2014/12/201412271243_529409_2960432_3.png 实验式:C10H18N4O4S3相对分子质量:354.5(按2007年国际相对原子质量计)生物活性:杀虫熔点:(173~174)℃蒸气压(20℃):5.1Pa溶解度:水中35mg/L,丙酮中8g/kg,甲醇中5g/kg,二甲苯中3g/kg。稳定性:60℃稳定,其水悬浮液因日光而分解,pH6稳定,pH9迅速水解,pH3缓慢水解。 1 范围本标准适用于硫双威及其生产中产生的杂质组成的硫双威原药。2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 1600-2001 农药水分的测定方法GB/T 1601-1993 农药pH值的测定方法GB/T 1604-1995 商品农药验收规则GB/T 1605-2001 商品农药采样方法GB 3796-2006 农药包装规则GB/T 6682-2008 分析实验室用水规格和试验方法GB/T 19138-2003 农药丙酮不溶物的测定方法HG 2611-1994 灭多威原药3 要求3.1 外观:白色至类白色固体。3.2硫双威原药应符合表1要求。表1 硫双威原药控制项目指标 项 目指 标硫双威质量分数,% ≥95.0灭多威质量分数,% ≤0.5水分质量分数,% ≤1.0pH值4.0~7.0丙酮不溶物质量分数,% ≤0.54 试验方法除另有说明,本试验所使用的试剂均为分析纯试剂,水应符合GB/T6682-2008中的三级水规格。4.1 抽样按照GB/T1605-2001中“商品原药采样”进行,用随机法确定抽样的包装件;最终抽样量不少于100g。4.2 鉴别试验 本鉴别试验可与硫双威含量的测定同时进行。在相同的色谱操作条件下,试样中某一色谱峰的保留时间与标样溶液中硫双威色谱峰的保留时间,其相对差值应在1.5%以内。4.3 硫双威质量分数的测定4.3.1方法提要试样用50%(V/V)四氢呋喃甲醇溶液溶解,以甲醇+水为流动相,使用Kroasil C18为填充物的不锈钢柱和可调波长紫外检测器,对试样中的硫双威进行高效液相色谱分离和测定。4.3.2试剂和溶液 水:新蒸二次蒸馏水,经0.45μm滤膜过滤;甲醇:色谱纯;四氢呋喃:HPLC; 硫双威标样:已知含量,≥99.0%。4.3.3仪器高效液相色谱仪:具可变波长紫外检测器;色谱数据处理机;色谱桩:250mm×4.6mm(id)不锈钢柱,内填Kroasil C18[
标定硫代硫酸钠时,是说等到溶液由红棕色变为淡黄色再加入淀粉指示剂,表示快到终点了,那我可以不加淀粉指示剂吗,直接滴到无色,这样对结果有影响吗?
如何配制间苯二酚硫酸溶液
异硫氰酸苯酯做为衍生试剂时,可以用分析纯的替代吗?我们刚要分析一种新的氨基酸,需要用异硫氰酸苯酯,可是到处都买不到色谱纯的,请问大家,可以用分析纯的代替吗??多谢多谢了.[em0808]
我这里是污水处理厂,以前出水的粪大肠菌群都是合格的,但最近两周一直超,想找一下是什么原因造成的!1.试想是硫代硫酸钠的问题:测同时间同漂水投加量同出水流量,测了三组数据,一组空白,一组采样品无硫代硫酸钠,一组采样瓶有硫代硫酸钠,添加量按标准,测出来的结果第一组未有阳性份数,小于20。第二组未有阳性份数,小于20。第三组结果为2800。2.猜想是次氯酸钠投加量不足造成的:同样次氯酸钠开最大,取3个水样,分别是出水流量8000.7000.6000,实验中其他因素一致,变量为出水流量,但检测出的结果分别为210,1100,310,三个水样中的余氯值分别为0.1,0.1,0.5。粪大肠菌群的结果没有呈现出规律性,水样中加了硫代硫酸钠仍检测出余氯,表示漂水投加量足够,样品中的余氯量超过了添加的硫代硫酸钠能抑制的量。3.猜想是水样检测时间的问题:同样猜想2中的3瓶水样,在冷藏保存16个小时后检测,检测结果未出,可预见的粪大肠菌群结果值比猜想2的更大。4.猜想是采样点位太近,漂水与出水未充分反应:这个猜想未做实验,同样的操作以前的水样都是小于20,但是最近的三次结果就高达9200
以铅为例: 1.原理:双硫腙与某些金属离子形成络合物容于氯仿,四氯化碳等有机物溶剂中,在一定的平H值下,双硫腙可与不同的金属离子呈现出不同的颜色,在加入掩蔽剂和其他消除干扰的试剂后调节平H=8.5-9.0时铅离子可与双硫腙形成双硫腙铅,可被三氯甲烷萃取出来,根据三氯甲烷呈现颜色与标准比色,540mm测定)。双硫腙可与许多金属元素反应,在周期表中可与20多种金属反应,所以我们就应该排除干扰离子,否则会影响测定效果。 2.排除干扰离子的方法有 ⑴调溶液的pH值(最理想的方法); ⑵改变金属离子的价数; ⑶加入掩蔽剂使干扰元素不与双硫腙反应,使干扰离子生成稳定的络合物。 对于这三种方法可同时使用,也可单独使用。理想的方法是两种以上配合使用。 用双硫腙法测Pb,双硫腙+Pb→形成络合物,这个实验的干扰离子有Fe3+、Sn4+、Cu2+、Cd2+、Zn2+等,为除去上干扰离子我们采用调pH=8-9进行掩蔽,而且KCN也不能在酸性中进行,这个实验的缺点就是用了大量的KCN(浓度20%),最低浓度为10%,又因为Fe3+ +3CN- →Fe(CN)3高铁氢化物,生成Fe(CN)3具有氧化作用,即可氧化双硫腙,为防止这一点我们加入NH2OH.HCL,盐酸羟胺使 Fe3+ → Fe2+ ,另外一般还加入柠檬酸铵进行掩蔽,加的目的是因为PH在碱性中,金属离子与碱生成金属离子的氢氧化物Mg(OH)2,所以加柠檬酸铵可以阻止Mg(OH)2的生成。 用双硫腙的关键是控制PH只有控制PH到8.5-9.0时才能加KCN,比色的波长选540mm,样品的处理采用湿法破坏(硝酸-硫酸法) 吸收处理后样10ml→于100ml的分液漏斗中→加20%柠檬酸铵2ml→20%盐酸羟胺1ml→酚红指示剂2d →用浓氨水调PH值8.5-9.0(颜色由黄→微红色)→加10%氢化钾1ml→ 摇匀→加双硫腙氯仿应用液10ml→摇动后分层→将氯仿层放入干净的10ml比色管中→于540nm下测定E→ 从标准曲线上查出相应的含量 3.计算: Pb(mg/kg)=V0*0.001*V2/W*V1 V0---样品消化后稀释的总体积(ml) V1---吸收消化液的体积(ml) V2---样品管相当于标准管的毫升数 W---样品重量(g)或体积(ml) 0.001---铅标准溶液的浓度(克) 1000---表示单位(重量1000克,体积1000ml) 4.注意事项 a. KCN是剧毒药品,操作时不能用嘴吸,使用后要洗手,KCN溶液不要与酸接触以防产生氢化氢气体而中毒。用下述办法可以降低氢化钾的毒性:向浓KCN溶液中加入氢氧化钠和硫酸亚铁使它生成餮氢化钾,降低毒性。 b. Pb和双硫腙结合其颜色由绿变为浅蓝→浅灰色→灰色→灰白→淡紫色→紫→淡红→红色 c. 本法测定重金属的灵敏度很高,在分析之前对所用的玻璃仪器要用1:3的稀硝酸浸泡,然后用水清洗干净。对高蛋白、高汤样品消化时,应先加硝酸缓缓加热待剧烈反应后,稍冷,再加入硫酸继续消化以防泡沫溢出。
二硫代氨基甲酸盐(或酯)类农药怎样定性?丙森锌代森铵代森联代森锰锌代森锌福美双福美锌在2763中有7种农药的残留物都是二硫代氨基甲酸盐(或酯),以二硫化碳表示。可以知道具体是哪一种吗?
关于四溴双酚A和六溴环十二烷的测定标准计划项目意见如何写?
薄层色谱法1 应用范围本方法适用于眼部化妆品及卸装品中防腐剂硫柳汞和苯基汞的定性测定。2 原理样品经浸提处理后,置检液于硅胶板上,经与双硫踪络合后,用己烷-丙酮液展开,根据与标准的Rf 值比较进行定性。3 试剂3.1 0.004%双硫腙氯仿溶液(1)。3.2 展开液:己烷十丙酮(90+10)。3.3硫柳汞标准溶液:称取相当含汞1.000g(2)的硫柳汞,溶于95%乙醇十水(1+1)溶液,补加95%乙醇十水(1+1)至100ml刻度。此溶液含汞10.0mg/ml。取一定量此溶液,用95%乙醇稀释至含汞0.35mg/ml的溶液。3.4苯基汞盐标准溶液:称取相当含汞1.000g的苯基汞盐于95%乙醇十水(1+1)溶液中,并稀释至100ml刻度,此溶液每毫升含汞10.0mg。取一定量此溶液用95%乙醇稀释至每毫升含汞0.35mg。4 仪器4.1 层析缸。4.2 高效硅胶薄层板:带富集区,Merekl3727或13728或等效物。5 分析步骤5.1 样品处理取5g样品分散于15m1 95%乙醇中,超声匀浆15min,用中速滤纸过滤,滤液在水浴锅上蒸发至近干,将残渣溶于lml 95%乙醇。5.2 分别取2.0μ1检液和标准点样于硅胶薄层板的富集区,每次1μ1。用一玻璃盖覆盖在硅胶薄层板上,但勿将富集区覆盖。将双硫腙氯仿溶液喷布在富集区上,如检液含有汞化合物,则与双硫腙形成络合物,将薄层板置于装有20ml展开液的层析缸内,盖上缸盖进行层析展开。展出液前沿移动6cm以后取出薄层板,观察橙色斑点的 Rf 值并与标准点进行比较,以判定检样中是否含有硫柳汞或苯基汞(4),以及其含量是否超过化妆品卫生标准限定最大用量(5)。
想用苯酚硫酸法测多糖,最近看了下但是还有些疑问,求大神指点。1如果苯酚没有变色是不是不用重蒸了,直接配成百分之5的苯酚溶液即可?2苯酚常温下不是固体麽,配制的时候直接称5克溶解定容100ml就可以了不?3一般说来百分之5是现配现用,但也有人说配制好放冰箱保存也可。我想问问,一般配制好的可以存放多久啊?4 加入苯酚后加入硫酸后,摇匀,在90℃的水浴中反应20分钟再在冰水中冷却5分钟。这里硫酸本身就会放热再放到沸水中。。。会不会爆沸啊







 我要推广仪器
我要推广仪器
 下载APP
下载APP