第六届中国蛋白质组学大会第一轮通知
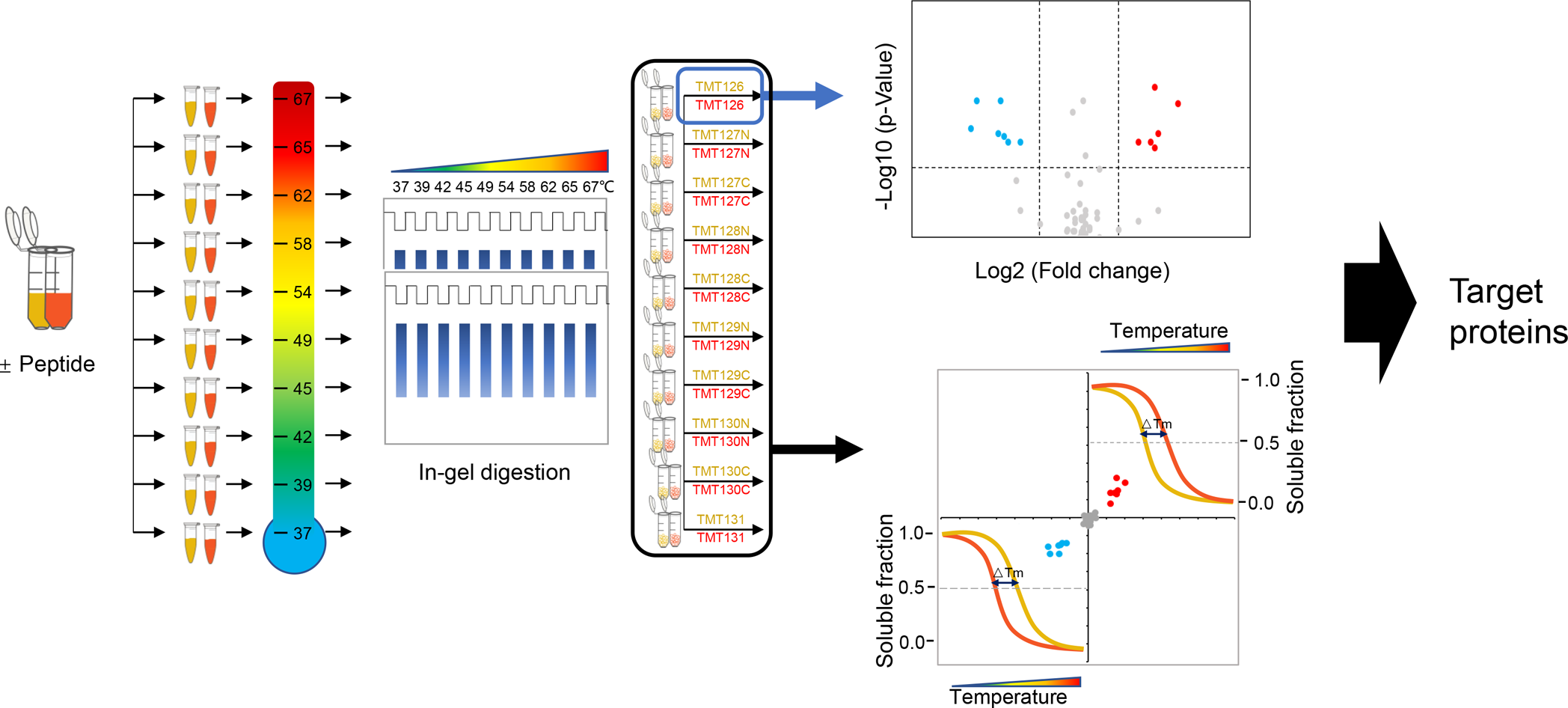
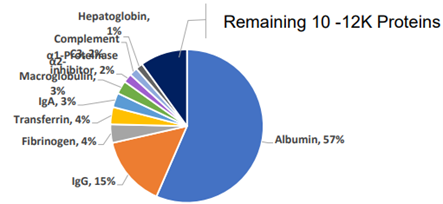
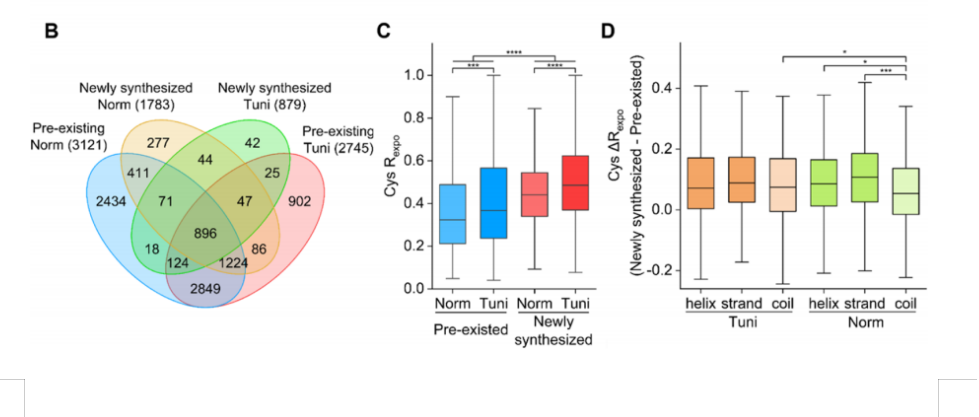
为了积极促进我国蛋白质组学的研究与发展,由中国生物化学与分子生物学会蛋白质组学专业委员会(CNHUPO)主办,北京蛋白质组研究中心和复旦大学共同承办的第六届中国蛋白质组学大会定于2009年7月28日—31日在江苏省泰州市召开。 一、会议安排 本届学术会议设有大会报告、分会(专题)报告和墙报三种形式。大会将邀请蛋白质组学及相关领域的国内外著名专家和教授作大会报告或专题报告,会议规模约600人左右。 会议同时举办与生物化学与分子生物学、蛋白质组学等研究领域相关的仪器、设备、试剂和新技术的展览、展示会。 大会安排于2009年7月28日举办蛋白质组学新技术培训,届时将邀请蛋白质组领域的国内、外知名专家授课。 二、会议议题 会议主要讨论蛋白质组学研究的现状及其进展,内容包括:疾病蛋白质组学,功能蛋白质组学,药物蛋白质组学,结构蛋白质组学,蛋白质化学,生物信息学,蛋白质修饰和相互作用,抗体相关技术,蛋白质组微分析、蛋白质芯片以及蛋白质组新技术新方法等研究领域。 三、会议语言 中文和英文 四、会议组织 组织单位 主办单位:中国生物化学与分子生物学会蛋白质组学专业委员会(CNHUPO) 承办单位:北京蛋白质组研究中心 复旦大学 主 席:贺福初院士 顾问委员会:(按姓氏汉语拼音顺序排列) 陈志南院士 强伯勤院士 饶子和院士 施蕴渝院士 汪尔康院士 王红阳院士 张玉奎院士 执行主席:钱小红 杨芃原 组织委员会 主 任:贺福初院士 副 主 任:杨芃原 杨晓明 钱小红 委 员:(按姓氏汉语拼音顺序排列) 陈志南院士 丁建平 高友鹤 何大澄 贺福初院士 李 明 梁宋平 刘斯奇 陆满晴 潘全威 钱小红 饶子和院士 施 前 王东根 杨芃原 杨晓明 杨秀荣 张先恩 张玉奎院士 甄 蓓 秘 书 长: 王东根 副秘书长:甄 蓓 施 前 学术委员会 主 任:饶子和院士 副 主 任:王红阳院士 张玉奎院士 委 员:(按姓氏汉语拼音顺序排列) 陈正军 陈志南院士 丁建平 高友鹤 何大澄 贺福初院士 李亦学 梁宋平 刘斯奇 刘银坤 钱小红 强伯勤院士 饶子和院士 施 前 施蕴渝院士 汪尔康院士 王红阳院士 杨芃原 杨晓明 张学敏 张玉奎院士 赵晓航 曾 嵘 甄 蓓 秘 书 长:钱小红 副秘书长:甄 蓓 施 前 秘书处 北京昌平区科学园路33号北京蛋白质组研究中心 电话:010-80705188 传 真:010-80705155 E-mail: cnhupo6@163.com 五、征文范围及要求(参照模版) 投稿论文收录入会议论文集,大会将组织优秀论文评选。参加会议代表将授予继续教育学分10分。 凡未在国内外公开刊物发表过的研究成果,均可投稿,具体要求如下: 征文范围:有关蛋白质组学及相关领域近年来研究的学术成果,以英文论文摘要形式投稿。 稿件要求:每篇论文摘要按正式发表论文要求撰写,限A4纸1页,使用Word软件撰写。文责自负。(参照模版) 字体要求:标题—Times New Roman四号加粗 作者—Times New Roman五号居中,拟作报告者请在其姓名下方划一横线。 注:大会报告幻灯片一律要求英文准备, 单位、地址、邮编、E-mail—Times New Roman小五号居中 摘要—Times New Roman五号 参考文献(Times New Roman五号)。 投稿方式:论文摘要请用E-mail附件传递,并在E-mail信中“主题”栏内写明“蛋白质组学会议” 若无上网条件请邮寄论文摘要一式两份,同时提交光盘(未提交者会议将不予录用)。 E-mail: cnhupo6@163.com 投稿地址:北京昌平区科学园路33号北京蛋白质组研究中心第六届中国蛋白质组学大会收 邮编:102206 截至日期:论文摘要投稿截至日期为2009年5月31日,以当地邮戳为准。 六、报到时间 2009年7月27日(参加培训的人员报到时间)、28-29日 七、会议注册费(国内代表) 2009年6月20日前注册:800元(人民币)/位(在读学生:500元/位) 2009年6月20日后注册:900元(人民币)/位(在读学生:600元/位) 技术培训费(与注册费一并交纳):100元(人民币)/位 八、重要时间提示 回执截止日期:2009年5月31日前(以当地邮戳为凭) 征稿截止日期:2009年5月31日前(以当地邮戳为凭) 会前注册时间:2009年6月20日前(以当地邮戳为凭) 九、交通指南 由于参会人员较多,大会组委会不安排车辆接送。请代表自行乘车抵达报到地点,望参会代表谅解。 十、注册须知 1.请与会代表携带本人身份证,学生代表需携带学生证。已交费代表请带好汇款凭证,以备核对。 2.注册代表权益: 正式代表和学生代表,可以参加会议组织的所有活动、注册费包括会务费、资料礼品费、会议安排旅游及7月29日—31日早餐、午餐和晚餐费用。 十一、会议地址和住宿宾馆 会议地址:江苏省泰州市扬子江药业集团海燕大酒店(江苏省泰州扬子江南路1号) 邮编:225300 住宿安排:海燕大酒店——标准间280元/天,单人间280元/天, 扬子江大酒店——标准间160元/天(距会场5分钟车程,会议期间班车接送) 十二、学术发言及证书登记 大会和分会发言及壁报交流的参会代表,请在注册当日(2009年7月27、28日)将报告材料或壁报资料(国际标准大小1.0m×1.2m)交至大会学术组,报告材料须为Powerpoint文件,一律要求英文准备,存储于移动硬盘、USB闪盘或光盘之中,大会提供笔记本电脑和幻灯放映设备,不接受个人电脑接入。如有特殊需求,请提前与大会学术组联系。大会发言鼓励使用英文,如有不便可使用中文。 论文被录用的参会代表可登记领取会议论文证书,所有参会代表可登记领取学分证书。 十三、旅游及定票须知 大会组委会设有旅游票务组专门负责参会代表的旅游及定票事宜。 组委会指定旅行社负责组织本次会议的会后(7月31日)旅游,会议期间请参会代表尽量不要安排旅游活动。旅游费用现场交纳并开具发票。军人和学生代表请携带相关证件,可享受门票优惠。 请参会代表至少提前两周提交返程票务预定单(附件二),否则组委会无法保证其预定。费用现场交纳,预订火车票须加收手续费,预订飞机票需向秘书组告知姓名及身份证号,不加收任何费用。 参会代表在注册报到时,须在旅游票务组登记确认自己的旅游和返程票务事宜,并交纳费用。会议期间如有问题,请随时与旅游票务组或大会秘书处联系。 十四、退费说明: 已交费的参会代表因个人原因不能参会或其他原因需要退款,请提前与会务组联系。退费原则:7月1日前退还所交款项的80%,7月1日~26日退还所交款项的50%,7月27日及以后恕不退款。(已预订火车票者扣除票价退票费和手续费后进行退费) 十五、联系方式 会议回执和论文发送或邮寄至: 第六届中国蛋白质组学大会会务组 电话:010-80705188 80705116 80705166 传真:010-80705155 E-mail:cnhupo6@163.com 地址:北京昌平区科学园路33号北京蛋白质组研究中心 邮编:102206 *请注明:“蛋白质组学大会” 注册费汇至: 帐 户:中国人民解放军62032部队 开户行:北京工商行永定路支行 帐 号: 0200004909008520585 * 务必注明:蛋白质组学大会*(汇款前请先打电话联系,汇款后将汇款凭据传真至我处,以确保汇款安全到帐) 十六 、附件 附件1:第一轮通知回执 附件2:返程票务预订单 附件3:论文摘要模板。 第六届中国蛋白质组学大会秘书处 二OO八年十二月一日 附件1 第六届中国蛋白质组学大会 第一轮通知 回 执 姓名: 性别: 职称: 联系电话: 工作单位: 传真: 通讯地址: 邮编: E-mail: 参加会议: 是 □ 否 □ 论文摘要题目: 所属专业:系统生物学□ 疾病蛋白质组学□ 功能蛋白质组学□ 药物蛋白质组学□ 结构蛋白质组学□ 蛋白质化学□ 生物信息学□ 蛋白质修饰和相互作用□抗体相关技术□ 蛋白质组微分析□ 蛋白质芯片□ 蛋白质组新技术新方法□ 其他□ 拟作报告形式: 大会报告 □ 分会报告 □ 墙报 □ 参加技术培训: 是 □ 否 □ 住宿标准: 海燕大酒店 (四星)标准间单人间 280元/天280元/天 扬子江大酒店 (三星)标准间 160元/天 选择房间类型: 海燕大酒店 标准间□ 单人间□ 扬子江大酒店 标准间□ 入住时间: 月 日~ 月 日 参加会议者请将回执于2009年5月31日之前发送或邮寄至: E-mail: cnhupo6@163.com 投稿地址:北京昌平区科学园路33号北京蛋白质组研究中心第六届中国蛋白质组学大会收 邮编:102206 附件2 返程票务预订单 返程日期航班号 (车次)目的地代表签字 备注 附件3 Identified the nonspecific binding proteins in depletion of Albumin and IgG from Human plasma Wang Yundan1, Ning Yunshan1,3, Jiang Yin2, Deng Xinyu2, Fang Qinmei2, Hong Yanhua3, Li Ming1,3 1 College of Biotechnology, Southern Medical University, Guangzhou, P. R. China, 510515 2 Beijing Institute of radiation Medicine, Beijing, P. R. China, 100850 3 Boang Antibody Company, Shanghai, P. R. China, 200233 tommy604@fimmu.com Depletion of high abundant proteins in plasma samples was necessary for the further study of new biomarkers mining in HPPP. We used the high specific mouse mAb against human albumin and Protein G to remove Albumin and IgG respectively from human plasma in denatured condition and native condition. We observed the different capacity of depletion in the presence of chaos reagents, non-ionic detergent and high concentration of salts. In native condition, the elution proteins were separated by 2DE and 104 spots in the gel were excised and trypsin digested for tandem mass spectrum (MS/MS) analysis. The binding proteins including Albumin, IgG, Fibrinogen, Vitamin D binding protein, Alpha-1 antitrypsin, transferrin, Transthyretin, Proapolipoprotein, Keratin, Complement component 3. The remained spots are albumin and IgG fragments. In denatured condition, the capacity of depletion for albumin become lower but IgG not affected. The concentration of nonspecific binding proteins including the fragments of Albumin in elution sample was lower. The results may explain the relation between low non-specific binding and presence of albumin fragments in condensed plasma samples processed by MARC or MARS system using commercial buffer. Keywords: High abundant protein / Depletion / 2-DE / MS / Nonspecific / Human plasma protein / Monoclonal antibody / Denature References 1. Huang, H. L., Stasyk T., Morandell, S., Mogg, M., et al., Electrophoresis 2005, 26, 2843-2849 2. Anderson, N. L., Polanski M., Pieper, R., Gatlin, T., et al., Molecular & Cellular Proteomics 2004 Apr 3(4):311-26. 3. Shen, Y. F., Kim, J. K., Strittmatter, E. F., Jacobs, J.M., et al., Proteomics 2005, 5,4034-4045 4. Omenn, G. S., States D. J., Adamski M., Blackwell T. W., et al., Proteomics 2005, 13, 3226-3245






















 我要推广仪器
我要推广仪器
 下载APP
下载APP