气相色谱-高分辨双聚焦磁质谱法检测,让血清无所遁形
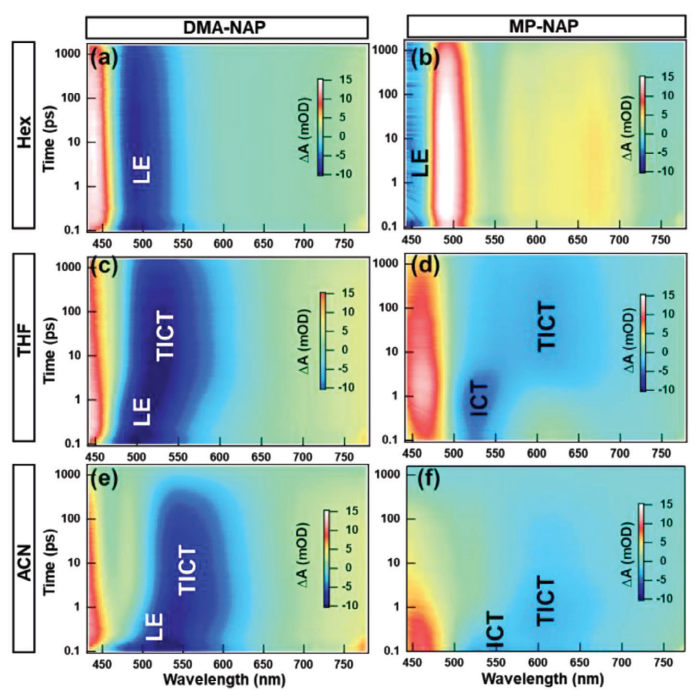
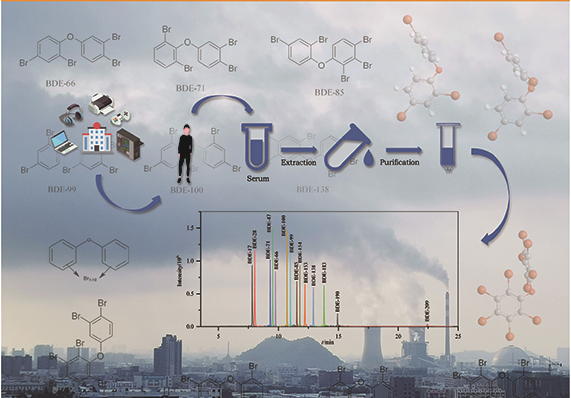
同位素内标-气相色谱-高分辨双聚焦磁质谱法检测血清中多溴联苯醚背景介绍 多溴联苯醚(PBDEs),是一种持久性有机污染物(POPs),根据苯环上溴原子的取代个数和位置的不同,共有10类209种同系物。由于其阻燃性能良好,被广泛应用于纺织品、玩具、建筑材料和电子设备等产品中。PBDEs的化学结构稳定,亲脂性强,容易释放到环境中,并通过食物链对生物体产生生物蓄积与生物放大作用,产生甲状腺毒性、神经毒性、内分泌毒性、生殖毒性、肝脏毒性、细胞毒性、致癌性等。 PBDEs对人体健康的影响已成为世界范围内高度关注的问题,目前针对多溴联苯醚人群暴露情况的研究,分析样本主要为血液、母乳和各种组织(脂肪、胎盘等)。由于多溴联苯醚是脂溶性化合物,在尿液中含量较低且多以羟基化代谢物的形式存在,脂肪组织的采样具有侵害性,且母乳和胎盘的采样仅限于一部分特殊人群,而血液样本相对较易获得,所以血液样本的测定是研究多溴联苯醚对人群健康影响的主要途径。 人体血清基质复杂,PBDEs含量较低,因此需提高富集效率并尽可能降低基质干扰,提高检测灵敏度。目前,液液萃取法、固相萃取法和加速溶剂萃取法是样品提取时较常使用的方法,样品净化主要使用凝胶色谱法和固相萃取柱净化法,检测方法主要有液相色谱-质谱法(LC-MS)、气相色谱-串联质谱法(GC-MS/MS)、气相色谱-负化学源质谱法(GC-NCI/MS)和气相色谱-高分辨双聚焦磁质谱法(GC-HRMS)。 LC-MS前处理步骤相对简便,但对PBDEs分辨能力较弱、灵敏度较低,更适合易热降解的高溴代多溴联苯醚的测定;GC-MS/MS、GC-NCI/MS选择性、灵敏度较高,对复杂基质抗干扰能力强,适用于痕量PBDEs的测定,但样本需求量较大,需采集2~5 mL血清样本;GC-HRMS同时备有静电场离子分析器和磁场质量分析器,因而使仪器同时具有能量聚焦和方向聚焦的双聚焦功能,灵敏度高、检出限低,适用于小体积样本中痕量和超痕量PBDEs的测定。 目前常用的GC-HRMS样品前处理步骤中主要采用凝胶色谱和酸性硅胶柱对样品进行净化,其中凝胶色谱法样本需求量较大(2 mL),酸性硅胶柱对实验人员填装操作要求较高,且无法同时测定多种PBDEs组分(如BDE-209等),批量样品检测时效率较低。 本方法探索使用少量血清(0.5 mL),采用GC-HRMS结合液液萃取和硅胶柱净化的方法,建立了人血清中14种PBDEs的测定方法,并用该方法对某地区15份青少年人群血样进行了检测,以期了解该地区青少年人群PBDEs的暴露水平。 样品前处理 血清样品解冻后移取0.5 mL于12 mL玻璃离心管中,分别加入200 μL硫酸、0.5 mL甲醇和20 μL内标使用溶液后混匀。先加入6 mL正己烷充分摇振后,以3500 r/min离心10 min,收集上层有机相;再加入6 mL甲基叔丁基醚,重复萃取,合并两次萃取液,于40 ℃、5 Pa氮吹25 min至0.5 mL。依次用2 mL甲醇和2 mL正己烷活化硅胶固相萃取柱,将浓缩液转移到硅胶柱上,先收集流出液,再用10 mL二氯甲烷-正己烷(1:1, v/v)溶液洗脱,合并流出液与洗脱液,40 ℃氮吹30 min至近干。向试管中加入10 μL正己烷复溶,振荡混匀,转移至棕色进样小瓶中,待测。 色谱条件 色谱柱:Rtx-1614毛细管柱(30 m×0.25 mm×0.1 μm);进样方式:不分流进样;进样口温度:290 ℃;传输线温度:320 ℃;升温程序:初始温度150 ℃,保持2 min,以15 ℃/min升温至250 ℃,保持1 min,再以25 ℃/min升温至290 ℃,保持3 min,然后以25 ℃/min升温至320 ℃,保持12.5 min;载气:氦气,恒定流量1.0 mL/min;进样量为1 μL。 质谱条件 电子轰击(EI)离子源,源温:280 ℃;电子能量:35 eV;电压选择离子检测(VSIR);分辨率:10000。14种PBDEs及其同位素内标的质谱参数见原文表1。 质量控制 样品前处理环境应在每次实验开始前和结束后进行清理,避免有目标物残留。实验过程中所用玻璃离心管、试剂、进样小瓶、固相萃取柱、枪头均做空白对照实验,未检出14种待测PBDEs。 文章信息 色谱, 2022, 40(4): 354-363 DOI: 10.3724/SP.J.1123.2021.10017 王梦梦, 谢琳娜, 朱英*, 陆一夫* 中国疾病预防控制中心环境与人群健康重点实验室, 中国疾病预防控制中心环境与健康相关产品安全所, 北京 100021



















 我要推广仪器
我要推广仪器
 下载APP
下载APP