[align=center][b][/b][/align][align=center][b][b]“嘌呤大户”被揪出!这4种食物才是痛风的祸源,再喜欢也别碰[/b][/b][/align]根据2021年数据显示,我国患有痛风这种疾病的人有1.466万人,超过了一千万人,而在这部分人群里,男性人群居多。随着人们生活方式和习惯的改变,更多人患上了痛风这种疾病,而很多人患痛风都是因为吃了大量的含有高嘌呤的食物。之但是如果患者的血尿酸水平居高不下,是不太建议食用含有嘌呤的食物的。所以,如果血尿酸水平比较高的时候,豆制品还是不吃比较好。如果已经患有痛风的人群,应该尽量不要食用含有高嘌呤的食物,以下4种食物,痛风患者不可食用。[align=center][b][b][font=宋体][color=#333333]第一种:啤酒[/color][/font][/b][/b][/align][color=#333333]有的人很喜欢喝酒,开心或者悲伤的时候,都喜欢喝一些酒来释放情绪,但是长期喝酒或者食用含有酒精类的食物,很容易引起痛风的发生。很多人觉得啤酒度数不高,喝点啤酒应该是没事的,如果这样想的话就大错特错了。啤酒是含有嘌呤比较高的饮品,饮用的话很容易引起血尿酸水平的升高,从而导致痛风的发生,或者加重痛风的病情,而如果痛风发作,还会增加患有其他疾病像是高血压,糖尿病的风险。所以,日常生活中,我们尽量少饮酒,痛风患者坚决不要喝啤酒,可以喝一些白开水或者其他不含或者含有嘌呤比较低的饮品来替代。[/color][b][font=宋体][color=#333333]第二种:动物内脏[/color][/font][/b][color=#333333]正常人吃了动物内脏对于身体不会造成什么不好的影响,但是痛风患者不能食用动物的内脏。因为动物内脏也是一种高嘌呤的食物,吃动物内脏会使得身体代谢紊乱,血尿酸水平升高,从而导致病情的加重。[/color][b][font=宋体][color=#333333]第三种:海鲜[/color][/font][/b][color=#333333]海鲜也是一种含有嘌呤比较高的食物,食用太多很容易诱发痛风。[/color][color=#333333]有些人在食用海鲜的时候,常常会搭配啤酒一起食用,这样食物中的嘌呤含量就更高了,如果是患有痛风的话,很容易对身体造成影响。[/color][color=#333333]所以,痛风患者像是生蚝、虾、螃蟹等等海鲜也是最好不要吃的,以免病情加重,出现身体不适。[/color][b][font=宋体][color=#333333]第四种:含糖饮料[/color][/font][/b][color=#333333]含糖饮料一般都会加入果糖来使饮料的口感更好,但是果糖在体内代谢之后会产生尿酸。[/color][color=#333333]而且还会导致尿酸的排泄的减少,长期喝果糖饮料,很容易患上痛风,而这种饮料也会加重痛风的症状,所以,日常生活中,不建议痛风患者饮用。以上就是我们关于四种嘌呤含量比较高会导致痛风的食物的介绍了,如果已经患有痛风,这些食物是都不建议食用的。[/color][color=#333333]同时,在日常生活中,痛风患者除了应该控制进食高嘌呤的食物之外,还应该注意饮食的搭配,适当地进行体育锻炼,劳逸结合,坚持进行药物的治疗,控制病情的发展。[/color]随着人们生活方式和习惯的改变,更多人患上了痛风这种疾病,而很多人患痛风都是因为吃了大量的含有高嘌呤的食物。[align=center][img]https://p0.ssl.img.360kuai.com/t01684c2d17b385a134.webp[/img][/align]根据2021年数据显示,我国患有痛风这种疾病的人有1.466万人,超过了一千万人,而在这部分人群里,男性人群居多。随着人们生活方式和习惯的改变,更多人患上了痛风这种疾病,而很多人患痛风都是因为吃了大量的含有高嘌呤的食物。之前有人说吃“豆制品”会导致痛风症状的加重,但其实虽然豆制品里边确实含有嘌呤,但是所含的并不多。[align=center]根据2021年数据显示,我国患有痛风这种疾病的人有1.466万人,超过了一千万人,而在这部分人群里,男性人群居多。[/align]随着人们生活方式和习惯的改变,更多人患上了痛风这种疾病,而很多人患痛风都是因为吃了大量的含有高嘌呤的食物。之前有人说吃“豆制品”会导致痛风症状的加重,但其实虽然豆制品里边确实含有嘌呤,但是所含的并不多。[img]https://p0.ssl.img.360kuai.com/t01cb0d14d26c0d7a6e.webp[/img]所以,如果患者的血尿酸水平比较稳定,少量地食用一些豆制品并不会对身体造成影响。但是如果患者的血尿酸水平居高不下,是不太建议食用含有嘌呤的食物的。所以,如果血尿酸水平比较高的时候,豆制品还是不吃比较好。如果已经患有痛风的人群,应该尽量不要食用含有高嘌呤的食物,以下4种食物,痛风患者不可食用。
用IC能做氯乙基磷酸吗?电导检测器。
求助:2-氯腺嘌呤的HPLC方法,先谢谢了!!
求助:2-氯腺嘌呤的含量测定方法,谢谢!
急!急!急!腺嘌呤及6-卞氨基嘌呤的测试方法
请教2-氯腺嘌呤的溶解度及HPLC分析方法,谢谢!
2-氯乙基氯甲基醚,CAS:1462-33-5,英文名称叫Chloroethyl chloromethylether,但是在网上和化工大辞典上都找不到它的相关物理化学性质,愁啊
最近遇到一个4-乙基-2,3-双氧哌嗪酰氯的问题,网上资料比较少,不知各位都做过这个吗?大家都用什么方法分析?主要是含量纯度方面的,最好能给点相关资料[em09510][em09510][em09510]附带一个..[~200024~]
总氯和游离氯的 N,N-二乙基-1,4苯二胺滴定法的空白和样品数据有没有同志有啊 急急急!!!!
衷心请教各位高手:1、我们单位即将购进安捷伦GC7890A,配了不同的柱子,为了缩短摸索的时间,想请问用什么类型的柱子做邻苯二甲酸二(2-乙基己基)酯和环氧氯丙烷效果会比较好?2、做这两个项目需要注意些什么?3、国标上做环氧氯丙烷用的检测器是FID,可是据做过的人介绍,好像用FID没办法做出那么低的检测限,不过用ECD或质谱可以做标准物质,但做水样就一直会有干扰峰出现,且跟目标物分不开,请问这干扰物是什么?有什么办法可以分离?衷心期待各位高手能帮我解答,感激不尽!
蔬菜的嘌呤含量与动物内脏、海鲜、肉汤等动物蔬菜的嘌呤含量与动物内脏、海鲜、肉汤等动物性食物相比,总体来说确实要低一些,但扁豆、芦笋、紫菜、豆苗等嘌呤含量相对较高,痛风急性发作期患者也要尽量避免吃,缓解期减少进食次数和进食量。性食物相比,总体来说确实要低一些,但扁豆、芦笋、紫菜、豆苗等嘌呤含量相对较高,痛风急性发作期患者也要尽量避免吃,缓解期减少进食次数和进食量。
豆浆中的腺嘌呤,鸟嘌呤,黄嘌呤,次黄嘌呤是如何产生,如何相互转化的?望老师不吝赐教
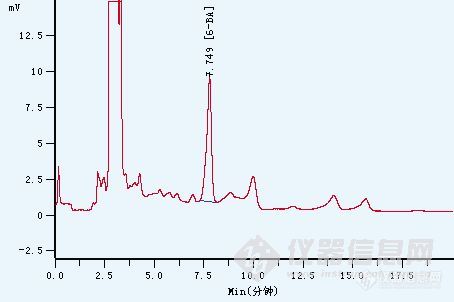
分享一个我公司实验人员优化的豆芽中的6-苄基腺嘌呤检测方法。豆芽中的6-苄基腺嘌呤检测,按照DB/11T 279-2006,北京市地方检测方法,用C18小柱富集,回收率只有40%左右,考虑到6-苄基腺嘌呤是酸性的物质,后来尝试了用CNW poly-sery MAX小柱富集,回收率可以做到80%以上。前处理方法:捣碎豆芽10g用20mL 酸化甲醇(甲醇/水=60:40,加入50μL乙酸)提取,离心,上清液旋转蒸发除去甲醇,调pH至9.0左右,待上样至固相萃取小柱。SPE固相萃取:活化:5mL 甲醇平衡:5mL 去离子水上样:流速 0.5滴/s淋洗:3mL 去离子水,3mL 20%甲醇水溶液,抽干洗脱:5mL 2%乙酸甲醇液相色谱条件:检测波长(nm): UV267nm柱型号: C18柱(4.6*250,5μm)流量(ml/min): 1.0mL/min流动相: 甲醇/1%乙酸=50/50柱温(℃): 30进样量: 20μL附上图谱(3ppm目标物)http://ng1.17img.cn/bbsfiles/images/2011/09/201109301339_320394_1776417_3.jpg
水质 游离氯和总氯的测定 N, N-二乙基-1, 4-苯二胺滴定法 HJ 585-2010标准简介:为贯彻《中华人民共和国环境保护法》和《中华人民共和国水污染防治法》,保护环境,保障人体健康,规范水中游离氯和总氯的检测方法,制定本标准。本标准规定了测定工业废水、医疗废水、生活污水、中水和污水再生的景观用水中游离氯和总氯的N, N-二乙基-1, 4-苯二胺滴定法。本标准是对《水质 游离氯和总氯的N, N-二乙基-1, 4-苯二胺滴定法》(GB11897-89)的修订。本标准的检出限(以Cl2计)为0.02mg/L,测定范围(以Cl2计)为0.08 mg/L~5.0 mg/L。对于游离氯和总氯浓度超过方法测定上限的样品,可适当稀释后测定。(注:原文见资料库)
本人最近在做关于腺嘌呤去氨基反应,到网上查找到重氮反应可以去氨基,但是做了几次都比较失败,所用的是在腺嘌呤中加入盐酸和亚硝酸钠,在0度左右进行反应,结果是腺嘌呤并没有太大变化,请问有做过腺嘌呤如何去氨基的吗,或者还有什么别的方法能去掉环上的氨基?
如题,本人想选择一款可以很好分离,成型四种嘌呤的液相色谱柱,分别是鸟嘌呤,腺嘌呤,黄嘌呤,次黄嘌呤,文献中的色谱柱是waters的Atlantic T3 流动相是0.05mol/L的磷酸二氢钾缓冲盐,不含有机相,现在我的疑问是1、waters这款柱子到底适不适合这种纯水相的条件2、waters这款色谱柱有点小贵,有没有其他品牌但是效果差不多的色谱柱呢,当然要是再贵一点,但是比这款柱子更适合的也可以希望懂的人能够给与解答,不胜感激
水质 游离氯和总氯的测定 N, N-二乙基-1, 4-苯二胺分光光度法 HJ 586-2010标准简介:为贯彻《中华人民共和国环境保护法》和《中华人民共和国水污染防治法》,保护环境,保障人体健康,规范水中游离氯和总氯的检测方法,制定本标准。本标准规定了测定地表水、工业废水、医疗废水、生活污水、中水和污水再生的景观用水中游离氯和总氯的N, N-二乙基-1, 4-苯二胺分光光度法和现场测定法。本标准是对《水质 游离氯和总氯的N, N-二乙基-1, 4-苯二胺分光光度法》(GB11898-89)的修订。本标准适用于地表水、工业废水、医疗废水、生活污水、中水和污水再生的景观用水中游离氯和总氯的测定,本标准不适用于测定较浑浊或色度较高的水样。对于高浓度样品,采用10mm比色皿,本方法的检出限(以Cl2计)为0.03mg/L,测定范围(以Cl2计)为0.12 mg/L~1.5 mg/L。对于低浓度样品,采用50mm比色皿,本方法的检出限(以Cl2计)为0.004mg/L,测定范围(以Cl2计)为0.016 mg/L~0.2 mg/L。对于游离氯和总氯浓度高于方法测定上限的样品,可适当稀释后测定。(注:原文见资料库)
GB/T5750.5-2006 N,N-二乙基对苯二胺分光光度法测定生活饮用水中游离氯,我用自制无需氯水做不出曲线,请问怎么可以准确配制无需氯水?用纯水代替无需氯水可以做出曲线,就是不知是否可代替,请各位大神帮帮忙
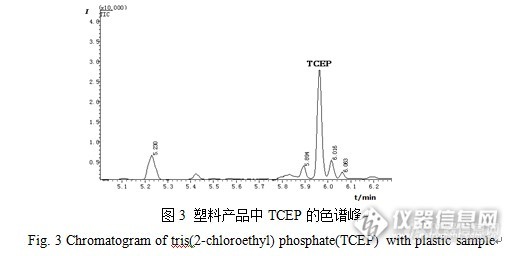
GC-MS法测定电子电气塑料产品中的磷酸三(2-氯乙基)酯 作者:陈梅http://ng1.17img.cn/bbsfiles/images/2015/12/201512071053_576565_2904170_3.jpg摘要:建立了气相色谱-质谱(GC-MS)法测定电子电气塑料产品中的有机磷酸酯阻燃剂——磷酸三(2-氯乙基)酯含量的分析检测方法。样品经微波萃取后,过固相硅胶净化小柱净化,然后用气相色谱-质谱仪进行选择离子监测模式下的定性及定量分析。结果表明:该方法的检出限为0.05 mg/kg,样品加标回收率为87.5%~92.9%,相对标准偏差小于4.1%。该方法适用于电子电气塑料产品中有机磷阻燃剂磷酸三(2-氯乙基)酯含量的快速测定,具有操作简单、方便、灵敏度高、结果稳定等优点。关键词:微波萃取;磷酸三(2-氯乙基)酯;电子电气产品;塑料;气相色谱-质谱法 磷酸三(2-氯乙基)酯(TCEP)是一种重要的有机磷阻燃剂,分子式为C6H12Cl3O4P,相对分子质量为285.49,化学结构式如图1所示。纯TCEP是一种浅黄色油状液体,微带奶油味,熔点为-64℃,沸点为194℃。常作为添加型阻燃剂广泛应用于建筑材料、电子电气产品的塑料部件、涂料、家具和纺织品等产品中。它具有阻燃效果持久,与聚合物基材相容性好,耐水、耐候、耐热以及耐迁移等特点。研究表明,TCEP阻燃剂化学性质很稳定,常温下难以降解,在体内具有生物累积性,长期接触能够侵害大脑组织,可以永久性地损伤记忆和学习能力,并且具有致癌性。研究表明,长期暴露TCEP可使F344大鼠出现致癌性及大脑退化损伤,随着暴露时间的延长,肝脏与肾脏重量明显增加,同时大鼠的死亡率也提高。因此,欧洲化学物质信息系统里明确将TCEP定义为致癌、生殖毒性和生态污染物,欧盟将其列入第二类高度关注物质,欧洲化学品管理局将其列入第二批授权物质清单。http://ng1.17img.cn/bbsfiles/images/2015/12/201512071054_576566_2904170_3.jpg 目前提取TCEP的主要技术有固相萃取、固相微萃取、超声波萃取、索氏提取和微波萃取等。检测方法有气相色谱法、气相色谱-质谱联用法和液相色谱-质谱法等。固相萃取及固相微萃取主要用于环境水样中TCEP的分析测试,索氏提取耗时长,一般需要6 h才能有效提取待分析物。杨左军等采用微波辅助萃取-高效液相色谱法测定电子电气产品中的含溴阻燃剂。然而,鲜有关于电子电气产品塑料部件中有机磷阻燃剂的检测研究报道。本实验以丙酮为提取剂,采用微波萃取法,建立了气相色谱-质谱(GC-MS)法测定电子电气塑料产品中TCEP的分析方法。1 实验部分1.1 仪器和试剂 气相色谱-质谱仪,QP2010 Plus,日本SHIMDAZU公司; 微波消解/萃取仪,Ethos ONE,意大利Milestone公司; 旋转蒸发仪,EV321,北京莱伯泰科仪器有限公司; 切割式粉碎机,SM300,德国RETSCH公司; 超离心粉碎仪,ZM 200,德国RETSCH公司; 超声波发生器,SB5200D,上海之信仪器有限公司; 氮吹仪,21011V001 R200,瑞士BUCHI公司; 电子天平,TB215D,美国丹佛公司。 磷酸三(2-氯乙基)酯(TCEP),纯度≥98.5%,德国DrEhrenstorfer公司; 丙酮,色谱纯,Sigma-Aldrich公司; 正己烷、二氯甲烷、乙酸乙酯、乙醇、乙腈均为分析纯,广州化学试剂厂。1.2 标准溶液配制 精确称取TCEP标准品0.100 g,用丙酮溶解并定容至100 ml,制备成1 000 mg/L TCEP标准储备溶液。临用前用丙酮将TCEP标准储备溶液稀释成50 mg/L,然后再逐级稀释成质量浓度分别为0.10、0.50、1.0、5.0、20 mg/L的标准工作溶液。1.3 气相色谱-质谱条件 气相色谱条件:SHIMDAZU Rtx-5MS色谱柱(30 m×0.25 mm,0.25 μm);升温程序:初始温度60℃(保持1min),以20℃/min升至260℃,再以20℃/min升至290℃保持1 min;载气为高纯氦气(纯度≥99.999%); 流速1.0 ml/min;进样口温度300℃;色谱-质谱接口温度280℃;进样量1.0 μL;进样方式为不分流进样;溶剂延迟时间3.5 min。 质谱条件:离子源温度230℃;四极杆温度150℃;采用电子轰击离子化(EI源)电离方式:电子能量70 eV;扫描方式:全扫描;扫描范围:50~350 amu;目标分析物定性离子的质荷比(m/z)为99.0、143.0、204.9和223.0,定量离子的质荷比(m/z)为249.0。1.4 样品处理根据电子电气塑料产品的不同成分特性,将其剪成小碎片,然后用切割式粉碎机粉碎制成2 mm左右的颗粒,最后用液氮冷却,经超离心粉碎仪粉碎成粉末状。准确称取0.500 g(精确至0.001 g)样品于微波萃取罐中,加入15 ml丙酮,按照选定的微波萃取条件进行萃取,萃取完成后,待萃取液冷却至室温,将萃取液转移至鸡心瓶中,并再用15 ml丙酮分三次洗涤萃取残渣,合并萃取液及洗涤液,旋转蒸发(控制温度低于45℃)至1 ml,再用氮气吹至近干,用1 ml丙酮溶解残渣并定容,再用0.45 μm有机滤膜过滤至样品瓶中,上气相色谱-质谱仪进行分析。若分析结果超过线性范围,可对萃取液进行稀释后再进行检测分析。2 结果与讨论2. 1 萃取条件的优化 分别用索氏抽提法和微波萃取法对含有TCEP的塑料样品进行提取,对测定结果进行比较,结果表明,索氏抽提6 h和微波萃取0.5 h的检测结果基本一致。考虑到微波萃取前处理方法简便、耗时少、试剂消耗少、可批量萃取等优点,因此,实验选用微波萃取法作为样品提取方法。2.1.1萃取剂的选择 采用微波萃取法提取样品时,不仅要考虑待分析物在溶剂中的溶解情况、溶剂与基质的相互作用,还要考虑微波吸收特性,溶剂的极性越强,对微波的吸收越强。分别采用正己烷、二氯甲烷、丙酮、乙酸乙酯、乙醇、乙腈6种萃取溶剂,对3款电子电气塑料产品进行萃取试验,结果如表1所示。从表1可以看出,在相同萃取条件下,丙酮的萃取效果明显好于其他萃取溶剂,因此,实验选择丙酮作为萃取溶剂。http://ng1.17img.cn/bbsfiles/images/2015/12/201512071058_576569_2904170_3.jpg2.1.2微波萃取温度和时间的选择 在微波萃取过程中,温度是重要参数之一,较高的萃取温度不仅有助于提高溶剂的溶解能力,降低溶剂的表面张力和黏度,而且还可以更好地破坏待分析物和基质活性部位之间的作用力,使目标物更易于从基质的活性部位脱附下来。同时,在进行微波萃取时,萃取罐内的压力也会随着温度的升高而升高,一般可达到几个甚至十几个大气压,压力升高导致萃取溶剂的沸点也随之上升,所以微波萃取时的温度可以比萃取溶剂的沸点高10~20℃,丙酮的沸点是56.48℃,所以选择最佳萃取温度为75℃。实验结果表明,采用梯度升温程序可有效、快速萃取电子电气塑料部件中的TCEP。一般情况下,微波萃取时间为15~20 min就可以保证达到很好的萃取效果,为了确保微波萃取待分析物完全彻底,实验选择整个萃取时间为30 min,具体萃取升
HJ 585-2010 水质 游离氯和总氯的测定 N,N-二乙基-1,4-苯二胺滴定法.pdf
求助,本人所在检测公司要做扩项,我要做HJ 586-2010水质 游离氯和总氯的测定 N,N-二乙基-1,4-苯二胺分光光度法的项目,要写方法验证报告,请问怎么写,在线等,急,谢谢大佬
HJ 586-2010 水质 游离氯和总氯的测定 N,N-二乙基-1,4-苯二胺分光光度法.pdf
哪位大侠有做过GB/5750.11-2006,方法是N,N-二乙基对苯二胺(DPD)分光光度法测定游离余氯或一氯胺的,可以交流一下吗,标准曲线3个9也好难做出来,小弟资质不够,能告之哪些步骤有干扰要注意的?或者开始线性不行,后来做好了,是怎么做好的?还有那个无需氯水是不是写错了,好像不好配的样子,我都用纯水代替了。
《水质 游离氯和总氯的测定,N,N-二乙基-1,4-苯二胺分光光度法》多次做标线,都达不到3个9,数据如下:ρ=0,A=0.004;ρ=0.10,A=0.007;ρ=0.20,A=0.028;ρ=0.30,A=0.049;ρ=0.50,A=0.093;ρ=1.00,A=0.189;ρ=1.50,A=0.274。请各位帮忙分析下可能是什么原因?试剂和操作都是按照标准进行的。谢谢!
黄嘌呤氧化酶(xanthine oxidase,XOD),可选择性催化氧化黄嘌呤和次黄嘌呤生成尿酸。有没有做过该酶传感器和对该酶性质了解的朋友?这种酶传感器似乎比较难做?因为:1. XOD活力小,0.67U/mg。2.检测对象次黄嘌呤在水中溶解度小,一般只能配到10^(-4)M级。所以电流响应始终做不出来。相同的方法换成葡萄糖氧化酶效果要好的多。大家多给意见。
一级水的电导率是多少
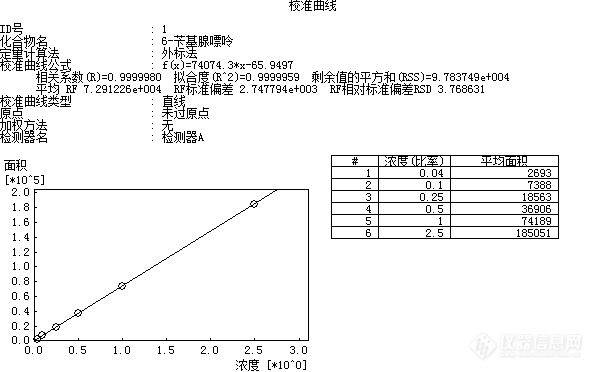
[align=center][b]食品中6-苄基腺嘌呤的测定方法验证报告[/b][/align][align=center][b]GB/T 23381-2009( 高效液相色谱法)[/b][/align][align=center][b]张霞[/b][/align]一、方法概述1.范围 本标准规定了用高效液相色谱法测定食品中6-苄基腺嘌呤(6-BA)含量的方法。 本标准适用于果蔬菜(豆芽、黄瓜、番茄、香菇、草莓、橙类)等植物性食品及其制品中6-苄基腺嘌呤的测定。2.规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方面研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法(GB/T 6682-2008,ISO 3696:1987,MOD)3.方法提要 试样经甲醇提取、浓缩并净化后,用高效液相色谱检测,外标法定量。二、仪器与试剂1. 仪器1.1高效液相色谱仪:配有紫外检测器或二极管阵列检测器。1.2 组织捣碎机。1.3离心机:转速不低于4000r/min。1.4超声波清洗仪。1.5旋转蒸发仪。1.6固相萃取装置。1.7电子天平:感量0.1mg。1.8微孔滤膜:0.45μm,有机相。以上仪器符合国标要求。2. 试剂及其配制 除另有规定外,所有试剂均为分析纯,水为GB/T 6682规定的一级水。2.1甲醇:色谱纯。2.2冰乙酸。2.3 C[sub]18[/sub]固相萃取柱:6mL,500mg,或相当者,使用前依次用5mL甲醇、10mL水活化。2.4乙酸铵溶液(0.02mol/L):称取1.54g乙酸铵,用适量水溶解,加入1.0mL冰乙酸,加水定容至1000mL。2.5 6-苄基腺嘌呤标准溶液(100.0μg/mL) [color=#ff0000] [/color][color=#ff0000]来源[/color][color=#ff0000]:[/color][color=#ff0000]农业部环境保护科研监测所[/color][color=#ff0000] [/color][color=#ff0000]货[/color][color=#ff0000]号[/color][color=#ff0000]:[/color][color=#ff0000]SB05-368-2016[/color][color=#ff0000] [/color]三、分析步骤1、标准曲线绘制1.1 标准工作液的配制: 分别吸取适量6-苄基腺嘌呤标准溶液,用甲醇定容至10mL容量瓶中,配制成浓度为0.04μg/mL、0.1μg/mL、0.25μg/mL、0.5μg/mL、1.0μg/mL、2.5μg/mL系列工作液。2、样品的处理2.1提取:称取经组织捣碎机捣碎的样品约10g(精确到0.01g)于50mL离心管中,加入20mL甲醇,超声提取15min,以转速不低于4000r/min离心10min,上清液转入50mL梨形瓶中,样品再次用20mL甲醇超声提取15min,离心合并上清液,用旋转蒸发仪(不超过60℃)浓缩至近干,去除甲醇,残液待净化。2.2纯化:将上述2.1残液以2mL/min流速通过预先活化的固相萃取柱,用少量水(约2mL)洗涤梨形瓶,洗液过固相萃取柱,再用5mL水洗涤固相萃取柱,去除杂质后用甲醇洗脱并定容至5.0mL,混匀后经0.45μm滤膜过滤,作为待测液供HPLC分析。3.仪器测定条件3.1色谱柱:C18柱,柱长250mm,内径4.6mm,粒径5μm或相当型号色谱柱。3.2流速:1.0mL/min。3.3柱温:30℃。3.4检测波长:267nm。3.5进样量:10μL。3.6流动相:甲醇 :0.02mol/L乙酸铵溶液=1:1四、结果处理试样6-苄基腺嘌呤含量按下式进行计算:[table][tr][td=1,2][align=center]X(mg/kg)=[/align][/td][td]C×[i]V[/i]×1000[/td][/tr][tr][td]m×1000[/td][/tr][/table]式中:X-试样中6-苄基腺嘌呤含量,单位为毫克每千克(mg/kg) C-由标准曲线计算出样液中6-苄基腺嘌呤的浓度,单位为微克每毫升(μg/mL) m-试样质量,单位为克(g) V-试样的最终定容体积,单位为毫升(mL)。1000—换算系数。计算结果保留两位有效数字。五、验证结果1.线性结果将标准系列工作溶液分别注入液相色谱仪中,测定相应的峰面积,以标准系列工作溶液的质量浓度为横坐标,以峰面积为纵坐标,绘制标准曲线。同时做空白实验。6-苄基腺嘌呤[u]Y=74074.3*X-65.9497 R^2=0.9999959[/u][align=center][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033263735_9846_2904018_3.png!w595x372.jpg[/img][/align]以上结果表明6-苄基腺嘌呤在0.04μg/mL~2.5μg/mL范围内,R[sup]^2[/sup]=0.9999959,6-苄基腺嘌呤浓度和峰面积呈线性关系,线性良好,符合要求。2.检出限结果将0.25μg/mL标准溶液逐级稀释至S/N=3±1,得出6-苄基腺嘌呤的方法检出限为0.0125mg/kg[color=#ff0000],[/color]此检出限结果小于国标GB/T 23381-2009的方法检出限0.02mg/kg,故此方法满足条件。六、方法精密度(重复性)对LBF180700282样品分别进行6次加标重复性的测定,测定结果如下:[table][tr][td][align=center]测定编号[/align][/td][td][align=center]1[/align][/td][td][align=center]2[/align][/td][td][align=center]3[/align][/td][td][align=center]4[/align][/td][td][align=center]5[/align][/td][td][align=center]6[/align][/td][/tr][tr][td][align=center]质量(g)[/align][/td][td][align=center]10.0031[/align][/td][td][align=center]10.0016[/align][/td][td][align=center]10.0025[/align][/td][td][align=center]10.0044[/align][/td][td][align=center]10.0027[/align][/td][td][align=center]10.0048[/align][/td][/tr][tr][td][align=center]浓度(μg/mL)[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.650[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.651[/align][/td][/tr][tr][td][align=center]含量(mg/kg) [/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.32[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][/tr][tr][td][align=center]平均值(mg/kg)[/align][/td][td=6,1][align=center]0.33[/align][/td][/tr][tr][td][align=center]RSD%[/align][/td][td=6,1][align=center]1.24[/align][/td][/tr][/table]本方法的精密度为1.24%,符合GB/T 23381-2009中给出试样测试结果的精密度要求。因此,本次测定均符合要求。七、准确度验证(加标回收)对LBF180700282样品加标,取2.5μg/mL的标液0.09mL、0.35mL、0.64mL同样品同步处理后,结果见下表:[table][tr][td=2,1][align=center]测定编号[/align][/td][td=6,1][align=center]6-苄基腺嘌呤[/align][/td][/tr][tr][td][align=center]序号[/align][/td][td][align=center]m(g)[/align][/td][td][align=center]V(mL)[/align][/td][td][align=center]C(μg/mL)[/align][/td][td][align=center]6-苄基腺嘌呤含量(mg/kg)[/align][/td][td][align=center]平均值(mg/kg)[/align][/td][td][align=center]加标量(mg/kg)[/align][/td][td][align=center]回收率%[/align][/td][/tr][tr][td][align=center]1#[/align][/td][td][align=center]10.0236[/align][/td][td][align=center]5.0[/align][/td][td][align=center]N.D[/align][/td][td][align=center]N.D[/align][/td][td=1,2][align=center]N.D[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][/tr][tr][td][align=center]2#[/align][/td][td][align=center]10.0157[/align][/td][td][align=center]5.0[/align][/td][td][align=center]N.D[/align][/td][td][align=center]N.D[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][/tr][tr][td][align=center]加标1#[/align][/td][td][align=center]10.0087[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.042[/align][/td][td][align=center]0.021[/align][/td][td][align=center]0.021[/align][/td][td][align=center]0.022[/align][/td][td][align=center]95.5[/align][/td][/tr][tr][td][align=center]加标2#[/align][/td][td][align=center]10.0103[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.163[/align][/td][td][align=center]0.081[/align][/td][td][align=center]0.081[/align][/td][td][align=center]0.087[/align][/td][td][align=center]93.1[/align][/td][/tr][tr][td][align=center]加标3#[/align][/td][td][align=center]10.0189[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.307[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.16[/align][/td][td][align=center]93.8[/align][/td][/tr][/table] 由上表可以看出6-苄基腺嘌呤测定的加标回收范围在 60%-120% ,RSD值为1.31%符合规定要求。八、总结从检出限、线性范围、重复性、回收率测试结果可知,均符合方法要求,本实验方法符合GB/T 23381-2009的要求。[img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img]
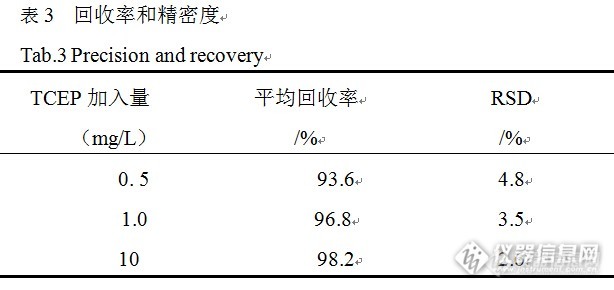
微波辅助萃取-GC-MS法测定玩具中磷酸三(2-氯乙基)酯 作者:蒋小良http://ng1.17img.cn/bbsfiles/images/2015/11/201511271307_575230_2904170_3.jpg 摘要:建立了微波辅助萃取-气相色谱-质谱法(GC-MS)测定玩具中磷酸三(2-氯乙基)酯含量的分析方法。考察了6种不同萃取溶剂对磷酸三(2-氯乙基)酯的萃取效率,选择了以乙酸乙酯为萃取溶剂,考察了微波辅助萃取温度与时间对萃取效果的影响,选择了最佳的微波辅助萃取条件。萃取液经浓缩后,用气相色谱-质谱仪进行选择离子监测模式下的定性及定量分析。结果表明,该方法的检出限为0.02 mg/kg,样品的加标回收率为93.6%~98.2%,相对标准偏差小于4.8%。该方法具有快速、方便、灵敏度高、定性准确等优点,适用于玩具中禁用有机磷阻燃剂磷酸三(2-氯乙基)酯含量的快速测定。 关键词:微波辅助萃取;磷酸三(2-氯乙基)酯;玩具;气相色谱-质谱法 近年来,由于溴代阻燃剂在全世界范围内逐步禁止使用,有机磷酸酯类阻燃剂作为其主要替代产品,其生产与使用得到了大幅度增长。所以,有机磷酸酯类物质也被广泛用于儿童产品(包括玩具)的阻燃整理工艺中,由此所引起的环境问题逐步引起了环境科学工作者的关注。有机磷酸酯类物质成为新型有机污染物研究又一个热点。有机磷阻燃剂化学性质很稳定,难以降解,具有生物累积性,能够侵害大脑组织,从而永久性地损伤人记忆和学习能力,并且具有致癌性,欧洲化学物质信息系统里明确将磷酸三(2-氯乙基)酯(tris(2-chloroethylphosphate), 简称TCEP)定义为致癌、生殖毒性和生态污染物。2010年1月,欧洲化学品管理局将三-( 2-氯乙基) 磷酸酯列入第二批授权物质清单;2012年,加拿大政府发布拟议法规修正案,将禁止在三岁以下儿童使用的产品(整体或部件)中使用含有磷酸三(2-氯乙基)酯的聚氨酯泡沫(PUF);欧洲报检和环境科学委员会(SCHER)就玩具中的TCEP表述科学观点,规定其限值为5 mg/kg,SCHER表示应该在玩具中禁用TECP及其同系物,此观点获得欧洲标准化消费者之声(Anec)和欧洲消费者联盟 (Beuc)的支持。为了更好的保护儿童健康和安全,欧委会接受相关组织的建议,发布2009/48/EC修订法案COM/2012/003,规定禁止在玩具中禁用TECP及其同系物;2011年7月,华盛顿生态部批准通过了《儿童产品安全法申报规则》第173-334-WAC章,该条例旨在收集相关信息,帮助政府和公众更好地了解儿童产品中潜在危险的化学品。该规则要求儿童产品生产商(包括品牌所有者和进口商)向生态部申报产品中对儿童具有高风险的化学品(CHCCs)的存在情况,其中TCEP已收录在需申报的化学品清单里。目前检测分析三-( 2-氯乙基) 磷酸酯主要方法有气相色谱法和气相色谱-质谱联用法。本文采用微波辅助乙酸乙酯萃取,气相色谱-质谱法(GC-MS)测定玩具中的三-( 2-氯乙基) 磷酸酯,对微波萃取条件进行了实验,选择了最近的萃取条件,并对该方法的精密度和加标回收率进行了系统研究,建立了玩具中三-( 2-氯乙基) 磷酸酯的快速测定方法。1、实验部分1.1 仪器和试剂 QP2010 Plus型 气相色谱-质谱仪 (日本SHIMDAZU公司);TB215D型 电子天平(美国丹佛公司);Ethos ONE型 微波消解/萃取仪(意大利Milestone 公司);EV321型 旋转蒸发仪(北京莱伯泰科仪器有限公司);ZM 200超离心粉碎仪(德国RETSCH公司);超声波发生器(上海之信仪器有限公司);21011V001R200型 氮吹仪(瑞士BUCHI公司)。磷酸三(2-氯乙基)酯(纯度≥98.5 %,DrEhrenstorfer公司),丙酮、乙酸乙酯、甲醇、乙腈、乙醇和正己烷均为色谱纯,均购自Sigma-Aldrich公司。磷酸三(2-氯乙基)酯标准储备溶液1000mg/L:精确称取磷酸三(2-氯乙基)酯标准品0. 10g,用丙酮溶解并定容至100 mL。临用前用乙酸乙酯将磷酸三(2-氯乙基)酯标准储备液稀释成50 mg/L的标准工作溶液。1.2 气相色谱-质谱条件 ( 1 )色谱条件:Rtx-5MS色谱柱( 30 m ×0. 25 mm×0. 25μm) ;升温程序: 初始温度70 ℃(保持2min),以20℃/min升至280 ℃,再以25 ℃/min升至290 ℃保持1 min; 载气: 高纯氦气; 流速: 1. 0 mL /min; 进样口温度280 ℃; 色谱-质谱接口温度:280 ℃;进样量: 1. 0μL;进样方式:不分流进样;溶剂延迟:5min。 (2)质谱条件:离子源温度230 ℃;四极杆温度:150℃;EI源:电子能量70 eV;扫描方式:全扫描;扫描范围:50~500amu。1.3 样品处理 取具有代表性的玩具样品10 g,剪碎至约为5 mm×5 mm,经超离心粉碎仪粉碎后(过1mm筛)。准确称取1 g(精确至0.001 g)样品于微波萃取罐中,加入15mL乙酸乙酯,按照选定的微波萃取条件进行萃取,萃取完成后,待萃取液冷却至室温,将萃取液转移至鸡心瓶中,并再用15mL乙酸乙酯分三次洗涤萃取残渣,合并萃取液及洗涤液,旋转蒸发(控制温度低于45℃)至近干,再用氮气吹至近干,用2mL乙酸乙酯溶解残渣并定容,再用0.2μm 滤膜过滤,滤液上气相色谱-质谱仪进行分析。2、结果与讨论2. 1 微波萃取条件的选择 影响微波辅助萃取效率的主要因素包括萃取溶剂种类和萃取条件,萃取条件主要有:萃取温度、萃取时间及萃取压力等。因此选择合适的萃取剂、萃取剂用量、萃取温度和萃取时间等,将直接影响样品中待测物萃取的效果及后续测试的结果准确度。2.1.1 萃取剂的选择 在微波辅助萃取中,选择萃取溶剂时,不仅要考虑样品中目标分析物在所选择溶剂中的溶解度,而且要考虑所选溶剂与样品基质的相互作用,以及所选溶剂对微波的吸收情况,溶剂的极性越大,对微波的吸收越强。按照样品处理程序1.3试验了二氯甲烷、乙酸乙酯、乙醇、甲醇、乙腈和正己烷6种萃取溶剂,对4种不同玩具产品进行萃取试验,实验结果见表1,由表1可见,在相同的萃取条件下,乙酸乙酯的萃取效率最高,所以实验选择乙酸乙酯作为萃取溶剂。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271317_575231_2904170_3.jpg2.1.2 微波萃取温度和时间的选择 微波辅助萃取温度是萃取过程中的重要参数之一,微波温度的高低将直接影响萃取速率和效率。试验研究表明,在进行
DPD(N,N-二乙基对苯二胺)指示剂加酸的原因是什么?是需要被激活才能显色吗?HJ 586-2010里面的硫酸可以换成其他酸吗?实验中配制的DPD指示剂总是变色,想换下配方。请各位大神答疑解惑!!!

在实验室做“HJ 586-2010 水质 游离氯和总氯的测定 N,N-二乙基-1,4-苯二胺分光光度法”方法验证时。多次绘制低浓度曲线都是这个走势(如下图)。整个操作过程都是按照标准细致操作,这是什么原因呢,请高手老师指导。或者有没有已经完成的低浓度曲线,可以借我参考下。http://ng1.17img.cn/bbsfiles/images/2010/12/201012271317_270049_2221461_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP