使用标准为LS/T3212-1992的挂面,领导说要检测铝含量,但是翻遍这个标准,没有一个字提到要检测铝含量,也没有引用标准说要检测铝含量?请问,要不要检铝?如果检应该适用那个标准?
用试剂盒方法(酶法)测定食品中葡聚糖含量摘要:描述了用试剂盒(酶法)测定食品中葡聚糖含量的方法前言: β-葡聚糖是禾谷类植物籽粒细胞壁中的多糖,是籽粒细胞壁的主要成分,化学名称为(1,3)(1,4) -β-D-葡聚糖,其含量以大麦和燕麦中较高其主要功能有:降低胆固醇,降血脂,调节血糖,提高免疫力,抗肿瘤和预防心血管疾病等。还有研究发现,它可以缓解和减轻肥胖症状。 β-葡聚糖测定方法包括酶测定法、荧光法、高效液相色谱法等,其中酶测定法因其反应专一性,测定准确可靠,而被采用为国际测定方法。酶法的方法过程如下:样品经水合和糊化,用地衣酶(β一葡聚糖酶)将样品中的β一葡聚糖酶解成β-葡基-寡聚糖。经调整体积和过滤分离后,用β一葡萄糖苷酶将这些可溶的B-葡基-寡聚糖水解成葡萄糖。葡萄糖用葡萄糖氧化酶氧化成葡萄糖酸和过氧化氢,后者用过氧化酶分解,以便在苯酚存在下,生色基4一氨基非那宗存在下形成适于比色分析的光吸收络合物,在紫外分光光度计下测定。 本文采用爱尔兰的Megazyme公司提供的测定(1,3)(1,4) -β-D-葡聚糖试剂盒,借鉴了EBC法3.11.1、AOAC法995.16,AACC法32-23方法,建立了测定相关制品中β-葡聚糖的测定方法。
联合国机构为牛奶中三聚氰胺含量设定新标准 根据世界卫生组织4日提供的消息,联合国负责制定食品安全标准的国际食品法典委员会为牛奶中三聚氰胺含量设定了新标准,今后每公斤液态牛奶中三聚氰胺含量不得超过0.15毫克。 国际食品法典委员会说,三聚氰胺含量新标准将有助于各国政府更好地保护消费者权益和健康。 国际食品法典委员会曾在两年前规定,每公斤用于制造奶粉的牛奶中三聚氰胺含量最多不得超过1毫克,其他食品中,三聚氰胺含量不得超过每公斤2.5毫克。 三聚氰胺是一种有机化学物质,广泛用于塑料、粘合剂、厨房台面、餐具等。曾有牛奶生产者向原料牛奶中掺入了水以增加体积。由于牛奶被稀释,牛奶中蛋白质含量降低。而使用牛奶进行下一步加工的公司,一般通过测量牛奶中含氮量来检测蛋白质含量。因此,添加三聚氰胺能提高牛奶的含氮量水平,从而造成蛋白质水平虚高。向食品中添加三聚氰胺从未获得过联合国食品法典委员会的批准。曾有食品生产企业非法将三聚氰胺添加到食品中,以明显提高蛋白质含量。 国际食品法典委员会系联合国粮农组织和世界卫生组织1963年联合设立的机构,专门负责协调政府间的食品标准,建立有关食品的国际标准体系。 专家称我国牛奶三聚氰胺含量比国际标准宽16倍 据《西雅图时报》今晨报道,世界卫生组织昨日称,联合国负责制定食品安全标准的国际食品法典委员会为牛奶中三聚氰胺含量设定了新标准,今后每千克液态牛奶中三聚氰胺含量不得超过0.15毫克。 该委员会说,新标准将有助于各国政府更好地保护消费者权益和健康。 新标准 不超0.15毫克/千克 禁人为添加 新标准规定,今后每千克液态牛奶中三聚氰胺含量不得超过0.15毫克。委员会曾规定,每千克用于制造奶粉的牛奶中三聚氰胺含量最多不得超过1毫克,其他食品中,三聚氰胺含量不得超过每千克2.5毫克。 世卫组织专家表示,三聚氰胺含量标准指食品中三聚氰胺自然的、不可避免的含量,而非人为添加的含量。制定三聚氰胺含量上限标准,有助于各国区别食品中无法避免且对健康无碍的三聚氰胺含量与蓄意添加三聚氰胺的行为。 向食品中添加三聚氰胺从未获得过联合国食品法典委员会的批准。 该标准不具备法律约束力。 国际食品法典委员会是联合国粮农组织和世界卫生组织1963年联合设立的机构,该机构由184个国家加欧盟地区的代表组成,专门负责协调政府间的食品标准,建立有关食品的国际标准体系。 小链接:三聚氰胺是一种有机化学物质,广泛用于塑料、黏合剂、厨房台面、餐具等。添加三聚氰胺能提高牛奶的含氮量水平,从而造成蛋白质水平虚高。 曾有牛奶生产者向原料牛奶中掺入了水以增加体积,导致牛奶中蛋白质含量降低。而使用牛奶进行下一步加工的公司,一般通过测量牛奶中含氮量来检测蛋白质含量。 乳业专家 国标比国际标准宽16倍 北京东方艾格咨询公司乳业分析师陈连芳上午接受《法制晚报》记者采访时表示,中国国家标准并不低于国际食品法典委员会的标准。他认为,把这样的标准在国际范围内合法化是一件好事。“从前是靠企业自觉,确立标准之后,一旦检测出三聚氰胺含量超标,便成了‘违规’。” 2011年4月20日,我国五部委就三聚氰胺问题发布公告,规定除婴儿配方食品中三聚氰胺限量值为1毫克/千克,其他所有食品(注:包括液态奶)均不得超2.5毫克/千克。 陈连芳表示,如此看来,我国2.5毫克/千克的标准远低于国际食品法典委员会新出台的标准。 “中国的标准更大程度上是临时限定,制定国标的原则就是参考国际食品法典委员会并考虑本国情况,相信中国的标准会根据实际情况进行修订。”陈连芳说。 2008年三鹿奶粉事件之后,欧美等国纷纷采纳了美国的“1毫克/千克”和“2.5毫克/千克”这两个指标,作为各国的规范,“中国也正是参考了这一数值,才制定了临时限量。”
用HPLC-ELSD分析单糖,双糖和寡聚糖时为什么流动相用乙腈,不用甲醇,不论用氨基柱还是carbohydrate的柱子都是用乙腈,仅有一部分用甲醇,但是相关用甲醇的文献在文中确实用乙腈或者水,一直在回避甲醇,不知为什么而且提取的时候也是用乙腈和水,都不用甲醇
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网[em0805]
[color=#0162f4] 联合国负责食品安全标准的机构国际食品法典委员会7月6日表示,该委员会已就食品中三聚氰胺的允许含量设立了新标准。新标准规定,每公斤婴儿配方奶粉中的三聚氰胺含量不能超过1毫克,而每公斤其他食品或动物饲料中的三聚氰胺含量不能超过2.5毫克。[/color][color=#000000] 世界卫生组织食品安全专家安格莉卡特里斯谢尔在当日的媒体吹风会上强调,所谓的三聚氰胺含量标准指的是食品中三聚氰胺自然的或者不可避免的含量,而非人为添加的含量。任何为了商业利益而故意在食品中添加三聚氰胺的行为都是不可接受的。有法可依了,好事![/color]
新华网日内瓦7月6日电(记者刘国远)联合国负责食品安全标准的机构国际食品法典委员会6日表示,该委员会已就食品中三聚氰胺的允许含量设立了新标准。新标准规定,每公斤婴儿配方奶粉中的三聚氰胺含量不能超过1毫克,而每公斤其他食品或动物饲料中的三聚氰胺含量不能超过2.5毫克。 世界卫生组织食品安全专家安格莉卡特里斯谢尔在当日的媒体吹风会上强调,所谓的三聚氰胺含量标准指的是食品中三聚氰胺自然的或者不可避免的含量,而非人为添加的含量。任何为了商业利益而故意在食品中添加三聚氰胺的行为都是不可接受的。 世卫组织食品安全、人畜共患病和食源性疾病部门负责人约恩施伦特则表示,食品卫生与安全问题是一个全球性问题,制定以科学为依据且所有国家都能遵守的共同标准非常重要。 国际食品法典委员会由联合国粮农组织和世卫组织共同建立,是一个以保障消费者健康和确保食品贸易公平为宗旨的制定国际食品标准的机构。该委员会本周在日内瓦召开第33届例会,主要讨论婴儿配方奶粉和其他食品中的三聚氰胺含量标准、猪肉中的瘦肉精残留以及蔬菜沙拉和海产品的卫生问题。
联合国机构为牛奶中三聚氰胺含量设定新标准2012年07月05日08:14 来源:新华网 新华网日内瓦7月4日电(记者 王昭 刘洋)根据世界卫生组织4日提供的消息,联合国负责制定食品安全标准的国际食品法典委员会为牛奶中三聚氰胺含量设定了新标准,今后每公斤液态牛奶中三聚氰胺含量不得超过0.15毫克。 国际食品法典委员会说,三聚氰胺含量新标准将有助于各国政府更好地保护消费者权益和健康。 国际食品法典委员会曾在两年前规定,每公斤用于制造奶粉的牛奶中三聚氰胺含量最多不得超过1毫克,其他食品中,三聚氰胺含量不得超过每公斤2.5毫克。 三聚氰胺是一种有机化学物质,广泛用于塑料、粘合剂、厨房台面、餐具等。曾有牛奶生产者向原料牛奶中掺入了水以增加体积。由于牛奶被稀释,牛奶中蛋白质含量降低。而使用牛奶进行下一步加工的公司,一般通过测量牛奶中含氮量来检测蛋白质含量。因此,添加三聚氰胺能提高牛奶的含氮量水平,从而造成蛋白质水平虚高。向食品中添加三聚氰胺从未获得过联合国食品法典委员会的批准。曾有食品生产企业非法将三聚氰胺添加到食品中,以明显提高蛋白质含量。 国际食品法典委员会系联合国粮农组织和世界卫生组织1963年联合设立的机构,专门负责协调政府间的食品标准,建立有关食品的国际标准体系。
[size=4][b][size=4]食品中三聚氰胺含量国际新标准出台[/size][/b][/size][size=3][b][size=4]2010-07-07 10:22 来源:新华网[/size][/b][/size][size=2][b][size=4][/size][/b][/size][size=4] 联合国负责食品安全标准的机构国际食品法典委员会6日表示,该委员会已就食品中三聚氰胺的允许含量设立了新标准。新标准规定,每公斤婴儿配方奶粉中的三聚氰胺含量不能超过1毫克,而每公斤其他食品或动物饲料中的三聚氰胺含量不能超过2.5毫克。[/size][size=4] 世界卫生组织食品安全专家安格莉卡特里斯谢尔在当日的媒体吹风会上强调,所谓的三聚氰胺含量标准指的是食品中三聚氰胺自然的或者不可避免的含量,而非人为添加的含量。任何为了商业利益而故意在食品中添加三聚氰胺的行为都是不可接受的。[/size][size=4] 世卫组织食品安全、人畜共患病和食源性疾病部门负责人约恩施伦特则表示,食品卫生与安全问题是一个全球性问题,制定以科学为依据且所有国家都能遵守的共同标准非常重要。[/size][size=4] 国际食品法典委员会由联合国粮农组织和世卫组织共同建立,是一个以保障消费者健康和确保食品贸易公平为宗旨的制定国际食品标准的机构。该委员会本周在日内瓦召开第33届例会,主要讨论婴儿配方奶粉和其他食品中的三聚氰胺含量标准、猪肉中的瘦肉精残留以及蔬菜沙拉和海产品的卫生问题。[/size]
联合国负责制定食品安全标准的国际食品法典委员会为牛奶中三聚氰胺含量设定了新标准,今后每公斤液态牛奶中三聚氰胺含量不得超过0.15毫克。这个指标应该是根据各类人群对三聚氰胺的耐受度而定的,中国人对此的耐受度呢?我国牛奶三聚氰胺含量比国际标准宽16倍,这不过是非常大的风险!实在相差很大!能放心饮用吗?牛奶企业真的符合国标吗?有多少是不合格的呢?
各位高手,我要用高效液相测定长链脂酰CAOs,需要一些关于C18的资料,以及固相提纯是用到寡聚核苷酸包被的柱子(The oligonucleotide purification cartridge),有没有人用过,那里可以买到,谢谢!
各位老师,最近用安捷伦的6495做了一批三聚氰胺样品,先跑的标准品,浓度为10、20、50、100、200 ng/mml再跑样品。大概跑了10个样品,我们又跑了一针10ng/ml的点,结果我们就发现后一针的含量明显低于10,只有2点多,整个跑样时间也很短,也就不到一个上午把。求各位大师,不吝赐教。感谢
用液相测大豆低聚糖含量,想问一下百分含量该怎样计算,百分含量相当于浓度吗,直接把峰面积带入标准曲线吗
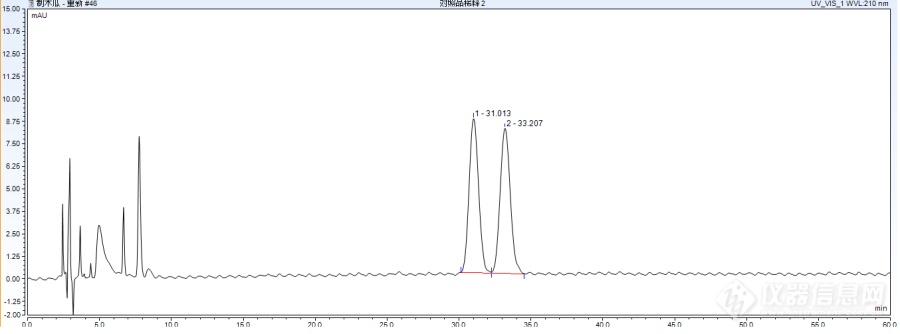
[align=center][b]制木瓜的含量测定研究[/b][/align] 制木瓜为蔷薇科植物贴梗海棠Chaenomeles speciosa(Sweet)Nakai 的干燥近成熟果实的加工炮制品。主产陕西、甘肃、四川、贵州、云南、广东。缅甸亦有分布。夏秋二季果实绿黄时采收,置沸水中烫至外皮灰白色,对半纵剖,切开,用酒蒸透后,晒干。本品呈类月牙形薄片,外表皮紫红色,有不规则的深皱纹。切面棕红色,气微清香,味酸。为了更好的控制制木瓜的质量,我们对制木瓜进行了含量方面的研究。为此,我们照高效液相色谱法(《中国药典》2015年版四部0512)测定,参照《中国药典》2015年版一部木瓜项下进行研究 1.[b]仪器与试剂:[/b]赛默飞ULTIMATE3000系列液相色谱仪(赛默飞公司);梅特勒-托利多分析天平([color=#333333]METTLERTOLEDO[/color]公司);超声波提取仪(上海精密仪器仪表有限公司);色谱甲醇(国药集团);自制超纯水;分析甲醇(国药集团)。齐墩果酸,批号:111522-200405,纯度:91.7%;熊果酸购自中国食品药品检定研究院,批号:111523-201208,纯度:93.8%[b] 2.方法与结果 2.1提取条件的选择:[/b]参照《中国药典》2015年版一部木瓜项含量测定方法中样品提取条件进行提取。[b] 2.2色谱条件与系统适用性试验:[/b]以十八烷基硅烷键合硅胶为填充剂;以甲醇-水 -冰醋酸-三乙胺265: 35:0.1:0.05)为流动相;检测波长为210mn。理论板数按齐墩果酸峰计算应不低于5000。 [b]2.3对照品溶液的制备:[/b]取齐墩果酸对照品及熊果酸对照品适量,精密称定,分别加甲醇制成对照品溶液,即得。色谱图见图1[img=,690,256]https://ng1.17img.cn/bbsfiles/images/2019/07/201907011738361809_7935_1839779_3.png!w690x256.jpg[/img] 图1 对照品的色谱图 [b]2.4供试品溶液的制备:[/b]取本品细粉约0. 5g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声处理20分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。色谱图见图2[img=,690,259]https://ng1.17img.cn/bbsfiles/images/2019/07/201907011739108378_6054_1839779_3.png!w690x259.jpg[/img] 图2 样品的色谱图[b] 2.5线性范围考察[/b]:精密称取齐墩果酸对照品18.10mg,分别制成每1ml含齐墩果酸16.598、33.195、49.793、66.391、82.989、165.977ug的溶液,各进样2次,每次10ul,测其峰面积,以峰面积平均值为纵坐标,进样量为横坐标,绘制标准曲线,得回归方程:Y=80.111X-0.0019,R[sup]2[/sup]=0.9995。结果表明齐墩果酸进样量在0.16598---1.65977ug范围内与峰面积呈良好的线性关系。精密称取熊果酸对照品20.15mg,分别制成每1ml含熊果酸18.901、37.801、56.702、75.603、94.504、189.007ug的溶液,各进样2次,每次10ul,测其峰面积,以峰面积平均值为纵坐标,进样量为横坐标,绘制标准曲线,得回归方程:Y=68.844X+0.0468,R[sup]2[/sup]=0.9995。结果表明熊果酸进样量在0.18901---1.89007ug范围内与峰面积呈良好的线性关系。[b] 2.6重复性实验:[/b]取ZMG-01-001样品6份,分别按供试品溶液制备方法处理,同法测定,样品中齐墩果酸含量分别为0.1423%,0.1414%,0.1520%,0.1412%,0.1411%,0.1485%,平均含量为0.144%,RSD为3.23%;样品中熊果酸含量分别为0.1681%,0.1619%,0.1723%,0.1637%,0.1716%,0.1709%,平均含量为0.168%,RSD为2.60%,[b] 2.7精密度试验:[/b]精密吸取齐墩果酸对照品和熊果酸对照品混合溶液10uL,按上述色谱条件连续进样5次,测得齐墩果酸峰面积分别是13.3963、13.2611、13.2801、13.1491、13.1934,峰面积RSD为0.71%(n=5);测得熊果酸峰面积分别是13.1621、12.9330、12.9726、12.9777、13.0619,峰面积RSD为0.70%(n=5),表明仪器精密度良好。[b] 2.8稳定性实验:[/b]取样品ZMG-01-003的供试品溶液,按上述色谱条件分别在0h,2h,8h,12h,24h依次进样,齐墩果酸峰面积分别是8.5416、8.3896、8.8179、8.9521、9.2791,齐墩果酸峰面积12小时内RSD为2.95%(n=4),齐墩果酸峰面积24小时内RSD为3.97%(n=5);熊果酸峰面积分别是2.2884、2.1677、2.2283、2.1758、2.4921,熊果酸峰面积12小时内RSD为2.52%(n=4),熊果酸峰面积24小时内RSD为5.86%(n=5),表明供试品溶液在12h内稳定。[b] 2.9加样回收率实验:[/b]取已知含量的同一样品ZMG-01-006的样品6份,每份取0. 25g精密称定,分别精密加入齐墩果酸和熊果酸对照品混合溶液4ml,按上述供试品溶液的制备方法和色谱条件测定,计算回收率,齐墩果酸平均回收率为96.12%,RSD为2.8%(n=6),熊果酸平均回收率为94.46%,RSD为2.9%(n=6)。结果表明,本法具有较好的回收率。(理论要求加样回收率90%~108%,RSD%5%)[b]样品测定:[/b]测定10批样品,根据数据计算参照《中国药典》2015年版一部木瓜项下制定,拟定含量限度为:不得少于0.40%(10批样品含量0.39%-0.82%之间。根据公式计算,限度应不得少于0.40%,结合《中国药典》2015年版一部木瓜项下的含量限度设定。)拟定本品含齐墩果酸(C[sub]22[/sub]H[sub]28[/sub]0[sub]11[/sub])和熊果酸(C[sub]22[/sub]H[sub]28[/sub]0[sub]10[/sub])的总量不得少于0.40%。[b] 3.讨论 3.1 [/b]关于指标成分的选择,我们参照2015版药典木瓜项下,确定熊果酸和其墩果酸作为含量控制的指标。[b] 3.2 [/b]本实验采用高效液相色谱法测定了制木瓜中的熊果酸和其墩果酸的含量,结果表明该方法准确,可靠。[b] 3.3 [/b]我们对10批样品中进行测定,结果不同产地的制木瓜含量有差别,可能与其生长环境有关。
SIGMA公司的标准品含量标识后有时写HPLC 或UV 不知是什么意思,另外标准品的含量是怎么得出的呢
联合国机构为牛奶三聚氰胺含量设新标准,今后每公斤液态牛奶中三聚氰胺含量不得超过0.15毫克。 其它食品为什么不跟随修改?是影响不大吗?
我现在要做一个工作标准品的含量,我们平时做一个含量,方法是等度,一个相关物质,方法是梯度的。我现在做标准品含量时两个方法都要做吗?做好了计算含量是用哪个方法的??
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=113155]《原料乳中三聚氰胺快速检测 液相色谱法》(GBT 22400-2008)国家标准.pdf[/url]附带:[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=113156]牛奶中三聚氰胺的含量测定.doc[/url]
来源:21世纪经济报道核心提示:目前,通过国家标准的《饮料通用分析方法》,我国只能对橙、柑、橘浓缩汁和果汁以及饮料,根据其果汁中可溶性固形物和6种组分实测值经计算后求得样品中的果汁含量,对其他类别果汁的含量则并没有可测定的标准。这意味着,产品中果汁含量是否标实相符,很多时候并没有办法核实。见习记者 曹晟源 上海报道 走入超市或是便利店,消费者很容易在果汁饮料的货架上看见100%果汁、纯果汁、纯果肉果汁等醒目的字样。而之所以会选择果汁饮料食用,买家主要是看上果汁较高的营养价值,这一营养价值正与果汁饮料中的果汁含量有着密切的联系,这也是消费者购买的重要依据。 在国内各大果汁企业生产的产品上,都会标注上5%或更高不等的果汁含量。 不过据记者了解,目前,通过国家标准的《饮料通用分析方法》,我国只能对橙、柑、橘浓缩汁和果汁以及饮料,根据其果汁中可溶性固形物和6种组分实测值经计算后求得样品中的果汁含量,对其他类别果汁的含量则并没有可测定的标准。这意味着,产品中果汁含量是否标实相符,很多时候并没有办法核实。 这就给一些果汁企业在生产过程中造成可乘之机,标注在多种果汁饮料背后的果汁含量数据,对于一些果汁企业来说很可能就仅仅是一个数字而已。 含量标准乱象:大多果汁无据可依 翻阅国家质量监督检验检疫总局和国家标准化委员会此前发布的《饮料通则》,记者发现,对于果汁(浆)在果汁饮料中的含量都有着一定量化的标准。 《饮料通则》中指出,果汁饮料中的果汁(浆)含量要大于等于10%,果肉饮料中的果浆含量不得低于20%,水果饮料中的果汁含量需在5~10%。 事实上,果汁饮料中的果汁含量也在一定程度上影响着消费者的购买行为。在价格相差不多的情况下,饮料中果汁含量较高的产品都会成为消费者的首选。但有意思的是,消费者一直依赖的这一选购果汁饮料的依据,很可能只是果汁厂家随意标注的一个数字。 据北京市质监局稽查大队执法室主任石海燕介绍,果汁或果汁饮料中果汁含量越高,意味着可溶性固形物含量就越高,而可溶性固形物含量通常用折光计测算所得的“折光度”来表达,所以企业生产中会参考折光度这项指标。 石海燕口中的“标准折光度”,是测定果汁含量的重要依据,但是这个重要依据从目前来看还有着一定的缺陷。 近日,记者就此采访了中国农业大学食品学院高彦祥教授,他表示目前中国的果汁行业除浓缩橙汁、橙汁及橙汁饮料果汁含量的测定有国家标准外,其他水果浓缩汁、果汁和果汁饮料果汁含量测定均没有国家标准,果汁企业均们是根据自己工厂制定的标准进行生产。 也就是说,目前在市面销售的除浓缩橙汁、橙汁及橙汁饮料果汁含量有据可依外,其他果汁饮料的标准却是一片乱象。 值得注意的是,即便是同一企业,在生产不同规格的同一种果汁产品时所制定的“标准折光度”也不一样。例如,有的企业在生产100%纯苹果汁时,内定按“标准折光度”10%的数值进行配料;而在生产苹果汁饮料时,则内定按“标准折光度”8%的数值进行配料。 石海燕指出,调查发现有的企业即使依据其自定的“标准折光度”计算,其产品的果汁含量也明显低于其标注的果汁含量,这就是涉嫌以次充好的故意行为了。这一问题不但在一些小型企业存在,在个别大型企业也存在。 不仅仅如此,国内更有果汁企业内部对于“标准折光度”都没有一个标准,其果汁饮料中果汁的含量就更加没有办法进行衡量。 标准的缺失背后,就可能导致果汁饮料包装上的果汁含量名不副实。换而言之正是由于没有相关国家标准进行严格的约束,厂家仅仅凭自律进行生产,在目前竞争激烈的果汁行业,难免出现以假充真、以次充好等违法行为。 检测标准亟待出台 高彦祥坦言,并不排除一些小型或是不负责任的果汁企业会利用法规漏洞、生产并不符合相关果汁含量的果汁饮料。“现在没有什么好的方法对此进行检测,因为目前连相关的标准都没有。” 因此,果汁饮料包装背后的果汁含量大部分并不具备真正的参考价值。而目前在零售环节,一瓶果汁饮料的价格都会按照果汁含量上涨,含量不小于20%的一瓶500ml的果汁饮料都要卖到五六块钱。但很可能事实上,这瓶果汁饮料的果汁含量并没有达到20%,差价中间利润的暴利或也由此出现。 标准的缺失,果汁含量的数据沦为摆设,有行业人士疾呼希望尽早结束这种没有约束的境况。 高彦祥指出,国家相关部门也在积极调研,希望能够尽快制定除橙、柑、橘以外,其他浓缩果汁、果汁和果汁饮料中果汁含量测定的标准,避免行业出现更多的质量问题。 目前,关于果汁含量检测有各种各样的设想,相关专家和机构提出的方案包括利用缓冲容量检测苹果汁饮料中果汁含量的方法、利用氨基氮含量及缓冲能力检测西番莲果汁饮料中果汁含量的方法、参照采用国外提出果汁饮料原汁含量的测定方法——RSK值(其中钾、硅酸盐的测定等效采用国际先进标准),以及建立饮料中总黄酮的测定方法等。 但是现实的难题还很多。中国地大物博,光是涉及苹果的品种就很多,每种苹果的质感不一,所得出的标准也不同,所以相关标准实现统一也尚有难度。 石海燕指出,虽然不能一次性解决所有的水果果汁饮料标准,但是国家有关部门应抓紧这一领域的技术攻关,从源头抓起,规范企业生产。尤其应对消费量最多的苹果汁、桃汁考虑优先制定标准。“科学准确地测定果汁含量不仅是企业组织生产的需要,更是政府实施有效监管,制止行业内不正当竞争的需要。”她说。 在她看来,当前条件下,“标准折光度”是目前行业内生产中判定果汁含量通行的简便方法。在浓缩果汁、果汁和果汁饮料的果汁含量测定标准制定前,制定“标准折光度”不失为一种替代方法。 “折光度标准的制定不仅影响到国内的企业生产、销售市场,还影响到我国果汁产品的国际竞争力,有关部门应结合我国的实际情况加以研究和完善,使之更加科学合理、简便易行,尽快制定发布我国果汁及果汁饮料的标准折光度行业标准。” 目前,国际果汁生产商联合会(IFU)都很重视食品法典和果汁生产标准的制定,国际食品法典委员会(CAC)已经对各种水果的折光度制定了标准,国内各企业在生产实践中也在摸索制定各自的“标准折光度”。
用液相测大豆低聚糖含量,样品出峰时间与标准品出峰时间不一致是什么原因
现在国家有没有标准规定,每kg液态牛奶中三聚氰胺含量不得超过多少mg?
解析我国与美国纤维含量测试标准的差异马志强摘要:综合概述了我国与美国在纤维含量分析标准上存在的主要差异,提供给生产、贸易和检测企业作为参考。关键词:纤维成分;美国;差异美国是我国服装类纺织品最主要的出口国,也是世界最大的纺织品消费市场,出口美国的产品必须要遵循美国本地的法令法规,特别针对纤维成分标签,美国有严格的标签法令,指导生产商和零售商进行正确的纤维标注。为了确保标签的正确性,首先要使用正确的测试方法。本文针对我国和美国在纤维含量测试方法上存在的差异进行了比较和分析,给国内的生产、贸易企业提供了相关的信息,使其能根据产品的出口地区,正确的进行产品纤维含量的分析和标签标注,满足市场的要求。1美国纤维含量测试标准的特点美国纺织染色家和化学家协会(AATCC)在纺织测试标准领域非常具有权威性,由其编制的针对纺织品的AATCC测试方法涵盖了纺织品纤维成份分析,色牢度实验及织物水洗的物理性能等测试标准。AATCC标准是纺织产品进入美国市场采用最为广泛的测试标准。AATCC测试标准在纤维含量测试方面有两个方法,AATCC20标准为纤维定性分析法;AATCC20A标准为纤维定量分析法。纤维定性分析中包括纤维纵向和横向截面显微镜观察法,燃烧法,密度法,溶解法,干捻法,粘色法,熔点法,红外光谱法。AATCC20A是纤维定量分析,包括显微镜分析法,化学溶解法和拆分法等。2我国标准与美国标准之间的差异我国对纺织产品的纤维含量测试是基于FZ/T 01057—2007《纺织纤维鉴别实验方法》和GB/T 2910《纺织品 定量化学分析》。在此之外还有行业协会和相关机构针对特殊产品编制的方法标准,如FZ/T 01095—2002《纺织品 氨纶产品纤维含量实验方法》、GB/T 16988—1997《特种动物纤维与绵羊毛混合物含量的测定》、FZ/T 30003—2009《麻棉混纺产品定量分析法》和FZ/T 01026—2009《定量化学分析 四组分纤维混合物》等。2.1纤维定性分析方法在所有检测机构中对纤维鉴别使用最为广泛的方法应该为显微镜观察法和溶解法,针对特殊的纤维还会辅助采用燃烧法、红外光谱法等,其他方法在实际检测过程中由于精准性和可操作性等原因,使用较少。现简单介绍几个定性分析方法的过程,以及差异。2.1.1显微镜观察法显微镜观察法依靠生物显微镜作为工具,通过技术人员直接观察纤维的纵向和横向截面,从而初步判断纤维的种类。一个经验丰富的技术人员,可以通过显微镜判断棉、麻、丝、毛等天然纤维,以及粘胶、氨纶等部分化学纤维。AATCC 20《纤维定性分析》标准和FZ/T 01057—2007《纺织纤维鉴别实验方法》,除了对每一种类纤维的纵向和横向外观形态进行语言描述之外,还提供了大量的纤维图片,给以更为直观的认知。2.1.2溶解法我国的行业标准FZ/T 01057.4—2007《纺织纤维鉴别实验方法 溶解法》要求一律采用常温(20℃~30℃)振荡5min,或煮沸3min之后观察样品的溶解情况。而AATCC20中溶解试验针对不同纤维溶解的难易程度提供了3种溶解时间,分别为5min,10min,20min。溶解温度除常温20℃之外还有50℃,90℃和煮沸。例如,二甲基甲酰胺法溶解确认试验,要求在90℃时处理10分钟之后再观察溶解现象。由于采用的溶解条件不同,某些特殊纤维的溶解现象会有所不同,检测人员应细心观察,注意区分。2.1.3干捻法AATCC20中用干捻法来初步判断麻类纤维。取平行纤维束,浸入水中,握住一端,使另一自由端对着观察者,在电炉上使用热空气加热,在纤维干燥的过程中,亚麻和苎麻纤维为顺时针旋转,大麻和黄麻纤维为逆时针旋转。2.2纤维定量分析方法GB/T2910—2009中除个别方法外均等同采用ISO1833最新版的方法,与美国标准体系有较多差异。美国AATCC20A标准中包含有含水率、非纤维物质去除、纤维含量拆分法、纤维含量化学分析法以及纤维含量显微镜分析法。而我国的纤维定量分析法根据方法的不同,有专门的标准与之对应。在进行国内纤维含量分析时要特别注意方法的选择,GB/T2910 为纤维定量化学分析法(附录中提供有手工拆分法),GB/T16988是针对动物纤维的显微镜分析法等等。2.2.1纤维定量化学分析法AATCC20A中提供了8种化学分析法:100%丙酮法、20%盐酸法、59.5%硫酸法、70%硫酸法、碱性次氯酸钠法、90%甲酸法、二甲基甲酰胺和二甲基乙酰胺法。而GB/T2910提供了23种化学分析法,覆盖了AATCC20A所有测试方法。现从样品准备、试剂选择、测试过程、数据处理等方面,分析两个方法体系之间的差异。1)样品准备(见表1)表1 备样方法比较步骤AATCC 20AGB/T 2910取样0.5~1.5g至少1g制样切成不大于3mm切成10mm左右烘干温度105~110℃(105±3)℃烘干时间至少1.5 h不少于4h,不超过16h冷却冷却至室温不少于2h称重精确至0.1mg精确至0.0002g恒重要求两次测量值在±0.001g无虽然在取样重量表述上有所不同,但是根据实际经验,两个标准的取样要求均能满足测试的代表性和准确性。在样品称重环节,AATCC明确要求样品烘干至恒重,并对恒重有明确的要求。而GB/T2910采用足够长的烘干时间,也能达到实际恒重的要求,但AATCC20A的可操作性更强些。2)试剂选择在进行相同的纤维含量分析过程中,AATCC与我国标准在溶解过程中试剂的选择有异同(见表 2)。表 2 部分方法的溶解试剂比较混纺类型AATCC 20AGB/T 2910醋酯纤维与其他纤维100%丙酮丙酮法聚酰胺纤维与其他纤维20%盐酸/90%甲酸甲酸法粘胶纤维与棉59.5%硫酸甲酸/氯化锌纤维素纤维与聚酯纤维70%硫酸硫酸蛋白质纤维与其他纤维次氯酸钠次氯酸钠腈纶、改性腈纶、氨纶和其他纤维二甲基甲酰胺/二甲基乙酰胺二甲基甲酰胺聚氨酯弹性纤维与其他纤维二甲基甲酰胺/二甲基乙酰胺二甲基乙酰胺[/td
有谁知道,测Polypropylene Glycol Methyl Ether (聚丙二醇甲醚)中聚合物含量的标准或是方法? 急啊~
因为cod含量偏低,所以想测测氯离子含量是多少?但是不知道测氯离子的含量的标准是哪一个,想要第三方检测公司在做的那一个标液,求大佬告知
我们化验室有气体吸收法和碳硫仪两种测试总碳含量的设备,想求购碳化钨载体的总碳含量在6.150到6.300±0.02的标准物质,最好是进口的,手头有货可以联系我
有关物质分析方法验证的可接受标准简介摘要:本文介绍了在对有关物质检查所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。 关键词:有关物质检查 分析方法验证 可接收标准 药品中的有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。由于这些有关物质的存在会影响到药品的纯度,进而可能会产生毒副作用,所以有关物质的控制是药品研发的一个重要方面,也是我们在药品审评中一直重点关注的要点之一。而要对有关物质进行严格的控制,就离不开专属性强、灵敏度高的分析方法,这就涉及到分析方法的筛选与验证。从现有的申报资料看,药品研发单位已基本上意识到分析方法验证的重要性,但是对验证时各具体指标是否可行尚没有一个明确的可接受标准,从而难以对验证结果进行评判。为解决这一问题,本文结合国外一些大型药品研发企业在此方面的要求,提出了在对有关物质检查方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。 1.准确度 该指标主要是通过回收率来反映。验证时一般要求根据有关物质的定量限与质量标准中该杂质的限度分别配制三个浓度的供试品溶液各三份(例如某杂质的限度为0.2%,则可分别配制该杂质浓度为0.1%、0.2%和0.3%的杂质溶液),分别测定其含量,将实测值与理论值比较,计算回收率,并计算9个回收率数据的相对标准差(RSD)。该项目的可接受的标准为:各浓度下的平均回收率均应在80%-120%之间,如杂质的浓度为定量限,则该浓度下的平均回收率可放宽至70%-130%,相对标准差应不大于10%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为:在定量限至一定的浓度范围内配制6份浓度不同的供试液,分别测定该杂质峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.990,Y轴截距应在100%响应值的25%以内,响应因子的相对标准差应不大于10%。 3.精密度 1)重复性 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于15%。 2)中间精密度 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于20%。 4.专属性 可接受的标准为:空白对照应无干扰,该杂质峰与其它峰应能完全分离,分离度不得小于2.0。 5.检测限 杂质峰与噪音峰信号的强度比应不得小于3。 6.定量限 杂质峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液杂质峰保留时间的相对标准差应不大于2.0%,峰面积的相对标准差应不大于5.0%。 7.耐用性 分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、检测波长变化±5nm、流速相对值变化±20%以及采用三根不同批号的色谱柱进行测定时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:各杂质峰的拖尾因子不得大于2.0,杂质峰与其他成分峰必须达到基线分离;各条件下的杂质含量数据(n=6)的相对标准差应不大于2.0%,杂质含量的绝对值在±0.1%以内。 8、系统适应性 配制6份相同浓度的杂质溶液进行分析,该杂质峰峰面积的相对标准差应不大于2.0%,保留时间的相对标准差应不大于1.0%。另外,杂质峰的拖尾因子不得大于2.0,理论塔板数应符合质量标准的规定。 9.溶液稳定性 按照分析方法分别配置对照品溶液与供试品溶液,平行测定两次主成分与杂质的含量,然后将上述溶液分别贮存在室温与冰箱冷藏室(4℃)中,在1、2、3、5和7天时分别平行测定两次主成分与杂质的含量。 可接受的标准为:主成分的含量变化的绝对值应不大于2.0%,杂质含量的绝对值在±0.1%以内,并不得出现新的大于报告限度的杂质。含量测定分析方法验证的可接受标准简介摘要:本文介绍了在对含量测定所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。关键词:含量测定 分析方法验证 可接收标准在进行质量研究的过程中,一项重要的工作就是要对质量标准中所涉及到的分析方法进行方法学验证,以保证所用的分析方法确实能够用于在研药品的质量控制。为规范对各种分析方法的验证要求,我国已于2005年颁布了分析方法验证的指导原则。该指导原则对需要验证的分析方法及验证的具体指标做了比较详细的阐述。但是文中未涉及各具体指标在验证时的可接受标准,国际上已颁布的指导原则中也未发现相关的要求。另一方面,大多数药品研发单位在进行质量研究时,已逐步认识到分析方法验证的必要性与重要性,大都也在按照指导原则的要求进行分析方法验证,但验证完后却因没有一个明确的可接受标准,而难以判断该分析方法是否符合要求。本文结合国外一些大型药品研发企业在此方面的要求,提出了在对含量测定方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。1.准确度该指标主要是通过回收率来反映。验证时一般要求分别配制浓度为80%、100%和120%的供试品溶液各三份,分别测定其含量,将实测值与理论值比较,计算回收率。可接受的标准为:各浓度下的平均回收率均应在98.0%-102.0%之间,9个回收率数据的相对标准差(RSD)应不大于2.0%。2.线性线性一般通过线性回归方程的形式来表示。具体的验证方法为:在80%至120%的浓度范围内配制6份浓度不同的供试液,分别测定其主峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。3.精密度1)重复性配制6份相同浓度的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于2.0%。2)中间精密度配制6份相同浓度的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于2.0%。4.专属性可接受的标准为:空白对照应无干扰,主成分与各有关物质应能完全分离,分离度不得小于2.0。以二极管阵列检测器进行纯度分析时,主峰的纯度因子应大于980。5.检测限主峰与噪音峰信号的强度比应不得小于3。6.定量限主峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液主峰的保留时间的相对标准差应不大于2.0%。7.耐用性分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、流速相对值变化±20%时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离;各条件下的含量数据(n=6)的相对标准差应不大于2.0%。8、系统适应性配制6份相同浓度的供试品溶液进行分析,主峰峰面积的相对标准差应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,主峰的理论塔板数应符合质量标准的规定。本文为转帖!
美国标准 测试金属中总镉含量 是那个标准?请教哈
产品质量标准中维生素含量范围值为:0.8×标示值~1.8×标示值,矿物质含量范围值为:0.75×标示值~1.25×标示值;产品中每种营养素含量的实测值必须在该产品质量标准范围值之内。营养素补充剂标示值等有关问题补充规定(征求意见稿)国家食品药品监督管理局药品注册司 二○○七年十月十日三、产品质量标准中营养素含量范围 (一)质量标准中维生素、矿物质含量范围依据标示值计算,并控制在标示值的80%~150%范围内。在SFDA网站上查不到《营养素补充剂审评规定》,应该是2004年SFDA的征求意见稿;《营养素补充剂标示值等有关问题补充规定》只是征求意见稿,应该是无效的;只有《营养素补充剂申报和审评规定(试行)》为有效的现行法规,是否就是应该按照《营养素补充剂申报和审评规定(试行)》1/3-2/3RNIS或AIS的原则进行确定?
请教各位高手,能用红外光谱仪测定乙丙丁三元共聚PP中的丁烯含量吗?据说先得有标准物,建立标准曲线才可以进行可靠的测量,但标准物也不好搞俺有个想法,即是目前化验室有现成的PE中丁烯含量测定方法,是否可以将已知丁烯含量的PE与均聚PP共混,然后测共混物中的丁烯与乙烯含量,然后建立标准曲线?







 我要推广仪器
我要推广仪器
 下载APP
下载APP