
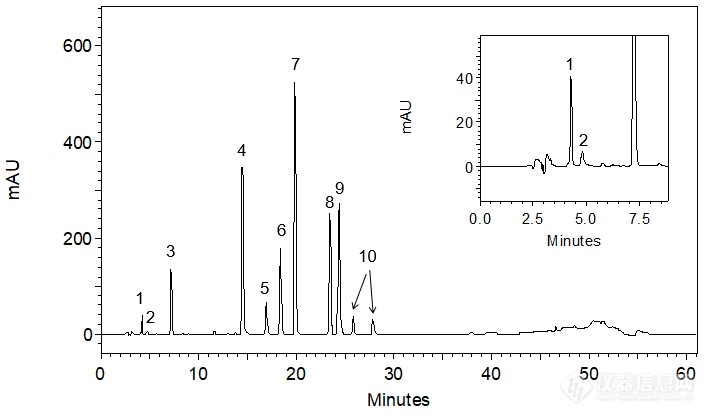
盐酸洛美利嗪含量测定方法研究本品为二苯哌嗪类钙通道阻滞剂,具有选择性的脑血管舒张作用。毒理研究遗传毒性:微生物回复突变试验、染色体畸变试验和小鼠微核试验结果均为阴性。下面主要针对盐酸洛美利嗪的含量测定方法进行研究。 一、容量法洛美利嗪为有机碱,可与高氯酸发生酸碱中和反应。1.指示剂选择和滴定终点的确定精密称取盐酸洛美利嗪约0.2g,加入15ml冰醋酸,振摇使溶解,加入5ml醋酸酐及5ml醋酸汞试液,加入1滴结晶紫指示液,并用电位计指示电位的变化,描绘滴定曲线。试验证明,当电位发生突跃时,溶液呈黄绿色。以高氯酸滴定液(0.1mol/L)滴定,并将滴定的结果用空白校正。每1ml高氯酸滴定液(0.1mol/L)相当于27.073mg的盐酸洛美利嗪。2.指示剂滴定法与电位滴定法含量测定的结果比较精密称取10份样品,每份约0.2g,加入15ml冰醋酸,振摇使溶解,加入5ml醋酸酐及5ml醋酸汞试液,加入1滴结晶紫指示液,其中五份做电位法滴定,另外五份做指示剂法确定终点,分别计算含量,数据见表1,从数据可知,电位法和指示剂法结果基本一致。用指示剂指示终点的三批样品的结果见表1。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251617_415393_2583865_3.jpg3.重复性试验及中间精密度试验三天内对同一批样品分别按80%、100%、120%三个水平各称取二份,指示剂滴定法测定其含量,结果见表2, 结果表明本法重复性及精密度较好。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251618_415394_2583865_3.jpg二、高效液相色谱法(HPLC)1.色谱条件及系统适用性试验(1)色谱条件:色谱柱:以十八烷基硅烷键合硅胶为填充剂(Xtimate C18),250×4.6mm,5um。流动相:甲醇-0.03mol/L磷酸氢二钾缓冲液(用磷酸调节pH4.0)(85:15),使用前经0.45μm有机滤膜抽滤并脱气。检测波长:225nm流速:1.0ml/min进样体积:20μl(2)系统适用性试验:精密称取干燥恒重的对照品约25mg置50ml量瓶中,用流动相溶解并稀释至刻度,摇匀作为贮备液。精密量取贮备液5.0ml置50ml量瓶中,精密量取20ml注入液相色谱仪,记录色谱图,连续进样6次,计算精密度。结果见表3。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251619_415395_2583865_3.jpg由试验结果可知,RSD小于1%,表明该色谱条件下精密度良好,系统适用性符合规定。2.线性关系精密称取干燥恒重的对照品约25mg置50ml量瓶中,用流动相溶解并稀释至刻度,摇匀作为贮备液。精密量取贮备液3.0、4.0、5.0、6.0、7.0和8.0ml置50ml量瓶中,用流动相稀释定容,摇匀作为溶液1、2、3、4、5和6,各精密量取20μl注入液相色谱仪。以标准溶液的浓度作为横坐标,色谱峰峰面积为纵坐标,绘制标准曲线。结果见表4。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251620_415396_2583865_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/12/201212251612_415391_2583865_3.jpg3.含量测定方法及测定结果精密称取本品适量,用流动相制成每1ml中约含50mg盐酸洛美利嗪的溶液,作为供试品溶液。另称取经恒重的对照品,同法制成每1ml中约含50mg对照品溶液。按前述高效液相色谱条件,分别量取对照品溶液和供试品溶液各20ml注入色谱仪,记录色谱图,按外标法计算含量。三批样品的HPLC法含量测定结果见表7-16。三批样品的含量测定结果见表5. http://ng1.17img.cn/bbsfiles/images/2012/12/201212251620_415397_2583865_3.jpg三、结果讨论分别采用容量法和高效液相色谱法测定三批样品的含量,可以看出两种方法的准确度、精密度等均能满足盐酸洛美利嗪含量检测的要求。其中容量法相对简单,系统误差小,故采用容量法作为含量测定的方法。
[align=center][b]头孢克洛有关物质——与9种杂质的共同分析[/b][/align]头孢克洛(cefaclor)为白色至微黄色粉末或结晶性粉末的化学品,微臭,本品在水中微溶,在甲醇、乙醇、三氯甲烷或二氯甲烷中几乎不溶,分子式:C15H14ClN3O4S。头孢克洛是β-内酰胺类抗生素,头孢菌素类药,是第二代头孢菌素,主要适用于敏感菌所致的急性咽炎、急性扁桃体炎、中耳炎、支气管炎、肺炎等呼吸道感染、皮肤软组织感染和尿路感染等。[align=center][img=,144,171]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140859582934_5220_2222981_3.gif!w144x171.jpg[/img][/align][align=center]头孢克洛[/align][align=center]M.W.: 367.81[/align]本实验对客户提供的头孢克洛原料药以及9种杂质(杂质A、B、C、D、E,7-ACCA,头孢克洛δ-3异构体,α-苯甘氨酸,苯甘氨酸甲酯盐酸盐)进行分析,希望得到杂质混合对照溶液及供试品溶液中各杂质的良好分离。客户反馈,将流动相磷酸盐体系的pH值由4.0提高到4.5可得到杂质混合对照溶液中7-ACCA和α-苯甘氨酸之间的良好分离,但头孢克洛与其相邻杂质E峰之间分离较难。客户前期使用了CAPCELL PAK C[sub]18 [/sub]MGII S3 4.6 mm i.d. × 250 mm色谱柱进行分析,在此基础上,我们尝试了其他填料的几款色谱柱进行分离尝试,分别为CAPCELL PAK C[sub]18[/sub] AQ(S3& S5)、CAPCELL PAK ADME(金刚烷基)、SUPERIOREX ODS、CAPCELL PAK PFP(五氟苯基)、CAPCELL PAK CN(氰基)。首先,参考客户提供的液相条件,使用高极性色谱柱[b]CAPCELL PAK C[sub]18 [/sub]AQ[/b]对杂质混合对照溶液进行分析尝试;为了得到杂质间的更好分离,粒径选择3 μm,如图1,[color=#2F5496]各杂质间均能得到良好的分离结果,头孢克洛与杂质[/color][color=#2F5496]E[/color][color=#2F5496]的分离度为[/color][color=#2F5496]2.70[/color][color=#2F5496],达到基线分离。[/color][color=#2F5496][/color][align=center][img=,690,405]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140902184290_9307_2222981_3.png!w690x405.jpg[/img][/align][align=center]图1 AQ S3 分析杂质混合对照溶液结果[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 头孢克洛 9. 杂质E [/color]10.杂质D[/align][color=#2F5496][img=,555,311]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140902187828_2715_2222981_3.png!w555x311.jpg[/img][/color]进一步分析供试品溶液,如图2,由于样品浓度较高,导致头孢克洛主峰向后展宽,进而将杂质E包于其中。[color=#2F5496][/color][align=center][color=#2F5496][img=,659,441]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140915544228_5404_2222981_3.png!w659x441.jpg[/img][/color][/align][align=center]图2 AQ S3 分析供试品溶液结果[/align][align=center][/align][align=left]为使头孢克洛和杂质E之间得到更好的分离,我们尝试对色谱条件进行调整。[/align][align=left][/align][align=left][b]1.调整柱温[/b][/align][align=left][b][/b]首先对温度进行调整:实验过程中发现柱温对头孢克洛与杂质E的出峰行为有较大影响——当柱温设置为20 ℃时,头孢克洛和杂质E之间能够得到良好分离;将温度提高到30℃时,杂质E向前移动趋势较大。为使杂质E峰出在头孢克洛峰前,避免由于供试品中头孢克洛峰的展宽而使杂质E被包于其内,进一步将柱温提高到40℃,发现头孢克洛与杂质E峰重合;最终,将柱温提高到45℃,此时杂质E峰移至头孢克洛峰前,但未能得到理想的分离结果。[/align][align=left][/align][align=center][img=,659,430]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140916597550_373_2222981_3.png!w659x430.jpg[/img][/align][align=center]图3 不同柱温条件下AQ S3分析杂质混合对照溶液结果[/align][align=center][/align][align=left][b]2.调整流动相[/b][/align][align=left][b][/b][/align][align=left]考虑到提高柱温对色谱柱寿命的影响,仍选择初始使用的20℃,对流动相梯度条件进行调整。在增强整体保留时间的同时,发现[color=#538135]头孢克洛和杂质[/color][color=#538135]E[/color][color=#538135]的出峰顺序发生了颠倒[/color],且[color=#538135]分离良好[/color],进而有效避免了杂质E被包于头孢克洛主峰中的问题;而在主峰后出峰的杂质D与头孢克洛之间分离度亦较高,即使供试品溶液中的头孢克洛峰展宽,也不会出现将杂质D包于其中的问题。[/align][align=left]因此我们在此梯度条件下进一步对供试品溶液进行分析,如图4,头孢克洛与各杂质峰之间均能得到良好的分离结果。[/align][align=left][/align][align=center][img=,679,417]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140917450308_6331_2222981_3.png!w679x417.jpg[/img][/align][align=center]图4 AQ S3分析杂质混合对照溶液及供试品溶液结果(调整梯度)[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 杂质E 9. 头孢克洛[/color] 10.杂质D[/align][align=left][img=,587,335]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918136074_9375_2222981_3.png!w587x335.jpg[/img][/align][align=left][/align][align=left]为使客户有更多的色谱柱选择,本实验室也尝试使用键合金刚烷基的高极性色谱柱CAPCELL PAK ADME分析杂质混合对照溶液和供试品溶液,如图5,在分析杂质混合对照溶液时,能够得到各组分的良好分离,同时发现杂质E和头孢克洛出峰顺序发生颠倒,但同时也发现头孢克洛峰与其后相邻杂质D峰之间分离度较低(Rs=1.71);因此,如图6,在分析供试品溶液时,由于色谱峰向后展宽,使得杂质D被包于头孢克洛主峰中,未能得到理想分离结果。[/align][align=left][/align][align=center][img=,690,426]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918484278_6616_2222981_3.png!w690x426.jpg[/img][/align][align=center]图5 ADME 分析杂质混合对照溶液结果[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 杂质E 9. 头孢克洛[/color] 10.杂质D[/align][align=left][/align][align=center][img=,689,417]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918485898_9906_2222981_3.png!w689x417.jpg[/img][/align][align=center]图6 ADME 分析杂质混合对照溶液结果[/align][align=left][img=,585,336]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140919331328_5070_2222981_3.png!w585x336.jpg[/img][/align][align=left][/align][align=left][/align][align=left]之后,我们也尝试使用了CN(氰基柱)和PFP(五氟苯基)以及高碳载量的SUPERIOREX ODS色谱柱,在客户提供的色谱条件下对杂质混合对照溶液进行分析,均未能得到更理想的分离结果。[/align]
公司作克林酶素磷酸酯,分析过程依据药典,使用ZOBARX C8分析,可分离效果很不理想.不知大家有没有做相同产品的,使用的什么柱子,最好有谱图,谢谢了.
一.事由 《新京报》2013年1月15日A30版体育新闻头条以《国乒接地气 马琳排第一》介绍了国乒史上首次网络票选结果:1.马琳 8250942.王皓 8206563.郝师 7709884.张继科 61万5.许昕 21万6.马龙 13万7.王励勤 不足2万投票:1月8日-1月14日票选前三将直接进入〝直通巴黎〞第三阶段终极选拔赛。二.网络票选分析化学家? 有可能吗? 我估计2015年或许会。时代在进步。三.如果我投票 1.汪尔康先生 2.俞汝勤先生 3.陈洪渊学兄 4.庞国芳先生 5.张新荣教授四.如果请你投票 1.你的投票标准认为应该包括那些呢? 2.刘国梁说:〝此次网络票选,只是一种游戏和尝试,但侧面也能反映出球迷对国兵队员的喜程度〞。 3.给您两年时间,现在是2013年。如果2015年请您投票,您将把票投给哪位分析家呢? 4.本文开头〝国乒接地气〞。相信只有接地气的分析化学家会获得分析化学工作者的网络选票。除非他∕她是真正的前瞻性分析化学研究,并也已证明是有用的。说明: 此文写於2013年1月15日
波谱及色谱联合应于小克银汉霉对氟比洛芬的生物转化作用研究 为了进一步减少对动物的测试过程,一些微生物如浮生真菌小克银汉霉和放线菌,已经证明对于外源性化合物的生物代谢方式类似于哺乳动物,早在30年前就有国外专家提出将微生物作为人体药物代谢的模型,虽然还有待进一步验证,但是微生物可以产生大量有用的药物中间代谢产物,他比从给药动物中分离这些化合物以及可能产生有毒物质或反应条件苛刻的的药物合成要可取的多。由于氟原子理想的化学性质(小的范德华半径,电负性,碳 - 氟键的强度),市场上大约25%的药物中含有氟原子。氟比洛芬〔(RS)-2-(2-氟-4-联苯基)丙酸〕就是一个这样的例子,它是一种非甾体抗炎药(NSAID),治疗关节炎引起的炎症。在人体它被代谢成Ⅰ相代谢产物4-羟基氟比洛芬,3,4-2羟基氟比洛芬,3-羟基-4-甲氧基氟比洛芬,以及葡萄糖酸和硫酸结合物(II期代谢产物)。在马的尿液中也检测到了羟基和甲氧基的代谢产物。尽管氟比洛芬药物应用普遍,但是对于其微生物的生物转化研究却很少。 材料与方法: 材料: 安捷伦高效液相、安捷伦气质联用仪、核磁共振波谱仪、旋转蒸发仪、恒温水浴锅、超声仪。 方法: 菌体在沙氏葡萄糖琼脂平板上在26℃下培养5天,然后均质化在100ml无菌生理盐水溶液中。均质浆接种在含有50ml新鲜沙氏葡萄糖肉汤的250ml锥形瓶中,28℃摇床孵育。将5mg的氟比洛芬溶解于20ul的二甲基甲酰胺中,72小时后加入到培养基中,继续培养5天,对照试验同样条件培养不加入的培养液。培养液冰浴超声10分钟,离心处理,上清液使用50ml的乙酸乙酯进行萃取,萃取液干燥 氟比洛芬代谢物的分析: 萃取物经Varian400-MHz核磁共振仪19F NMR谱表明氟比洛芬经三天培养后完全降解到为代谢产物。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281045_525026_2206101_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281045_525027_2206101_3.jpg 代谢产物的分离使用安捷伦制备液相,配18 9.4mm× 25cm色谱柱(Agilent Technologies),流动相采用20%至60%的乙腈进行梯度洗脱,流速4ml/min。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281047_525028_2206101_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281047_525029_2206101_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281047_525030_2206101_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281047_525031_2206101_3.jpg 结果与讨论: 1、对小克银汉霉对氟比洛芬的生物转化途径进行了如下推到:http://ng1.17img.cn/bbsfiles/images/2014/11/201411281049_525032_2206101_3.jpg 2、本实验中我们对小克银汉霉对于氟比洛芬的生物转化代谢产物通过核磁共振测定(NMR)的波谱,气相色谱 - 质谱(GC-MS)和高压液相色谱(HPLC)进行了分析鉴定。

http://ng1.17img.cn/bbsfiles/images/2011/06/201106140856_299721_2185349_3.jpg6月9日,各媒体爆出这样一则新闻:《洛克沙砷在美暂停销售——加在饲料中使鸡体中含致癌物我国早已上市》。报道指出:经美国食品和药物管理局(FDA)鉴定,一种在禽类中使用的药物“洛克沙砷” (Roxarsone,依照我国《饲料药物添加剂使用规范》,后文统一称为“洛克沙胂”)可能致使鸡体内含有致癌物质砷,于是辉瑞公司决定暂停在美销售。
请问钙镁指示剂(固体)的,分析纯的一般是多少克一瓶呀?
设备名称: 型号 类别 型号 类别 E4406A 发射机测试仪 68347C 信号源 HP83752 信号源 54147A 网络分析仪及配件 D3000A 信号源 HP8752C 网络分析仪及配件 R3765 网络分析仪及配件 R3131 频谱分析仪 MG3670B 信号源 HP8594E 频谱分析仪 HP8753D 网络分析仪及配件 HP8720D 网络分析仪及配件 HP8510B 网络分析仪及配件 HP8561E 频谱分析仪 HP8563E 频谱分析仪 MS8604A 频谱分析仪 HP8648C 信号源 E4418A 功率计 MT8801C 无线通信测试仪 HP8510C 网络分析仪及配件 89441A 其它 E4436B 信号源 53132A 通用仪器 R3767CH 网络分析仪及配件 37247C 网络分析仪及配件 R3767CG 网络分析仪及配件 R3765BH 网络分析仪及配件 R3267 频谱分析仪 频谱分析仪/噪声仪/网络分析仪及配件/功率计/信号源 发射机测试仪/无线通信测试仪 其它/通用仪器 租 赁 商 品 型号 类别 HP8753D 网络分析仪及配件 R3765BH 网络分析仪及配件 公 司 简 介 美国易克来博(ELECTROLAB)电子测量仪器公司位于科技前沿阵地─硅谷,于1993年成立。 公司定位:及时向业界提供重新检修的电子测试仪器的公司。 主要产品:以HP、TEK、ADVANTEST、ANRITSU 等著名品牌的射频、微波、通讯测试仪器为主,始终坚持严格的质量标准,所有仪器均按原厂指标校验 公司承诺:交货迅速、品质可靠、价格合理、服务一流! 更多访问: http://www.electrolab.com.cn/

10,抽取5个版友);中奖名单:千层峰(注册ID:jxyan)大川之子,纵横四海(注册ID:chuangu120)m3071659(注册ID:m3071659)莫名其妙(注册ID:moyueqiu)sunpengwjh(注册ID:sunpengwjh)http://ng1.17img.cn/bbsfiles/images/2016/09/201609281518_612452_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609281518_612453_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================美洛昔康片方法:HPLC基质:药品应用编号:101386化合物:美洛昔康固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil C18(2) 5u 150 x 4.6mm样品前处理:【有关物质】 取本品细粉,加碱性甲醇溶液(取40%甲醇溶液100 ml,加0.4 mol/L氢氧化钠溶液6 ml,混匀)溶解并稀释成每1 ml中约含有1 mg的溶液,滤过,取续滤液作为供试品溶液;精密量取1 ml,置100 ml量瓶中,用上述碱性甲醇溶液稀释至刻度,摇匀,作为对照溶液。色谱条件:检测器:UV 270 nm 流动相:0.1 mol/L 乙酸铵溶液-甲醇(50:50) 进样量:20 ul文章出处:P611关键字:美洛昔康,2010版中国药典,HPLC,含量测定,钻石二代,Diamonsil C18(2)谱图:http://www.dikma.com.cn/Public/Uploads/images/meiluoxikang.GIF
亲,最近我在做小试,需要分析奥美拉唑氯化物的分析方法,HPLC条件是怎么样的。。。
【网络讲座】:禽肉和禽蛋产品中的抗生素检测技术 【讲座时间】:2016-12-13 10:00【主讲人】:曹冬,毕业于北京化工大学制药工程专业,北京市海淀区疾病预防控制中心理化检验科 检验师, 从事食品安全理化检验工作5年,主要研究方向为食品中生物毒素,兽药残留等,在该行业积累了丰富分析检测经验。同时为清华在读硕士研究生。【会议简介】近年来随着食品安全持续被社会各界所关注,对相应的食品安全检测方法层出不穷,技术不断进步。作为食品安全分析最具挑战的样品前处理技术,也在不断进步。安捷伦科技也在紧跟应用需求,不断开发新的产品及应用,为客户提供完备的解决方案。本次网络讲堂将会介绍安捷伦科技近期推出的禽类产品,包括鸡肉和鸡蛋中金刚烷胺、利巴韦林等兽药残留分析全面解决方案。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名参会:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/2210 4、报名及参会咨询:QQ群—290101720,扫码入群“食品”http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669539_2507958_3.gif
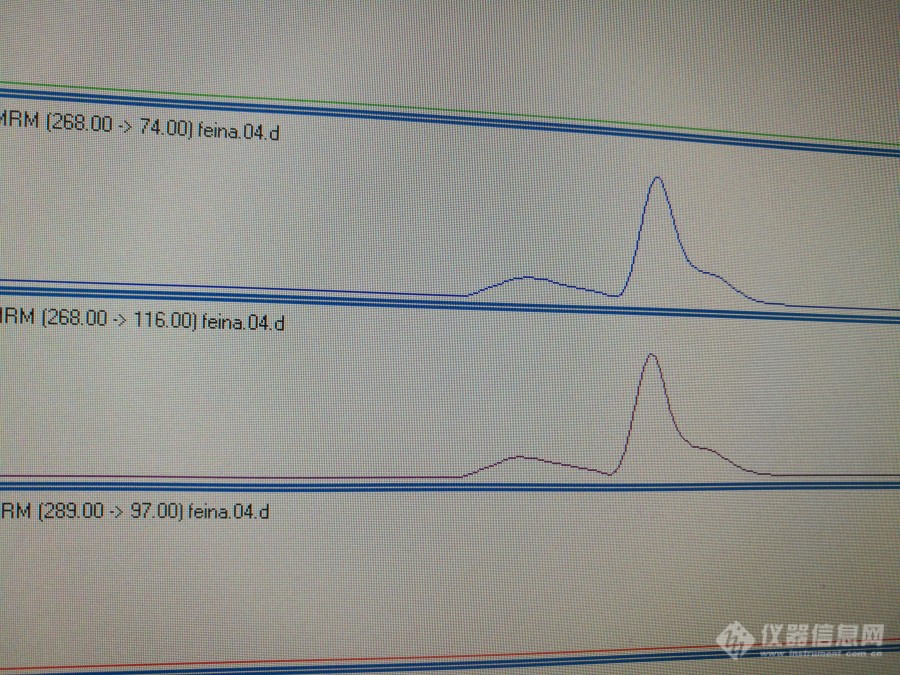
安捷伦6410QQQ采集的数据,对美托洛尔进行MRM,出现了图一中的双峰,改变流动相比例及PH值,峰型有所改变,但依然很差(见图二)。初始流动相,当有机相比例增大时(大于50%),峰型改善(也不好),后面的几种物质无法分开;当有机相比例偏小时(小于50%),则出现峰型极差或者图一中的双峰。请大家帮忙分析一下,谢谢![img=美托洛尔提取离子图-1,690,517]http://ng1.17img.cn/bbsfiles/images/2018/07/201807141707307198_1436_3440419_3.jpg!w690x517.jpg[/img],[img=美托洛尔提取离子图-2,690,517]http://ng1.17img.cn/bbsfiles/images/2018/07/201807141707402828_1450_3440419_3.jpg!w690x517.jpg[/img]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=152855][url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]法测定猪肝中克仑特罗残留的不确定度分析.pdf[/url] 测量不确定度是衡量测试结果准确性和可靠性的重要参数,测量结果的可用性很大程度上取决于其测量不确定度的大小,测量结果的表述必须同时包含被测量的值及与该值相关的测量不确定度,才是完整并有意义的。本文以猪肝中克仑特罗的残留测定方法为例对[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]在兽药残留测定方面的不确定度进行研究,建立了一套合理、完整的评定方案。
变化节奏快,在竞争十分激烈的行业和环境下,借助几颗one平台,企业可以实时解决“实时敌情”分析中最为重要的环节-----把握全局,把握当下。 某国内金融公司,因为没有及时了解到同行的投放策略,导致3天内,销售下降25%,损失近20万RMB。商场就是战场,市场竞争格局每几分钟都是不同 有没有办法每隔30分钟或者20分钟甚至更短的时间内,就能了解同行的市场投放策略,让自己在这每20分钟,自己能主宰自己的行业,或者在这个自己所在的行业灵活应变呢? 几颗one平台试图帮助客户解决这1个痛点,让其每1刻钟就能及时、实时调整自己的策略,避免错过最佳时机,同时又能全局全面对市场更有获得感。几颗one平台支持企业实时及时如每隔15分钟,分析同行的动作---如投放策略、排名策略、区域策略、关键性策略、价格策略、预算策略、市场趋势、全国布局策略、账户整体策略、新近同行策略、新退同行策略、早中晚营销策略、同行客服布局策略等,并能够通过多个角度对这些策略进行分析检测和以数字为评估。 具体而言,几颗one平台开发了一套自动追踪系统,可以实时及时对全国各地区监测同行对手的关键词百度的竞价排名,分析记录其排名为基础的各项参数。经过监测和分析,几颗one平台会把全国市场对手的重点变化和实时排名自动记录和发送给客户定制的渠道。比如,一家金融公司就产品价格在关键词排名进行了调整,并且计划今天及时抢占市场,几颗one平台将在每隔开15分钟,每循环15分钟内把金融公司的排名推送给客户,作为决策参考。 如果几颗one平台需要每隔15分钟监测数千个或者上万个变化,用户是通过什么办法分辨重要的变化呢? 还是以金融公司的竞争对手为例,在15分钟内,几千或者上万的关键词排名变化了,而百度搜索引擎的排名在各个地区的排名变化都会展示出来,比如,对手的关键词变化了,对手的策略变化了,都能看出来,那么对手一天的营销策略,都能及时体察到,如此一来,即使同行对手每隔15分钟调整自己的网络营销排名策略,都能及时了解到,并且能做出合适的平衡应对策略。 实际上,市场上专门提供类似工具的也有,但是能够对比全局市场的,并且能实时提醒到相关操作人员和相关市场投放人员,同时提醒市场管理人员实时知道同行策略,能对同行的变化实时的做出对应投放策略,针对同行的不同地区,不同排名,不同类型的关键词,及时收集,及时处理信息,为企业快速做出策略而给出全局沙盘战略信息,在几颗one系统中,几颗one系统解决的问题是:实时能做到15分钟级别或者更短时间的监测 沙盘能做到全局战略性 动态能及时了解同行的排名,不会出现同行变化了投放策略不能及时知道的情况 现在几颗one系统,拥有的客户中,大部分都是所在行业的前5名,也只有有一定经济实力的才配这个高度市场营销显微镜,几颗one系统的收费目前不低,只是简单投放市场关键词排名的就没必要了解这个了,如果需要对同行及时关注排名和同行排名变化的动态的,那么可以尝试试一试。 几颗one的竞争对手也有,如第三方的网络服务公司,社会化的自媒体监测公司等。但是几颗one系统任务,相比主要从事专业信息动态处理,几颗one的优势在于,专门围绕竞争同行的百度竞价排名进行及时、实时、自动提醒到相关环节,让同行实时“排名情报”更有可决策性和平衡性和针对性。
急需苯嗪草酮分析方法,那位帮帮忙啊?不甚感激!
http://ng1.17img.cn/bbsfiles/images/2017/01/201701191656_647918_2507958_3.gif梅特勒热分析在线技术交流会主讲人:李焱,现任梅特勒公司热分析仪器部技术应用顾问活动时间:2013年10月24日 下午 14:00http://ng1.17img.cn/bbsfiles/images/2017/01/201701191656_647918_2507958_3.gif【简介】 主作为全球热分析技术领域的领导者,梅特勒-托利多公司一直致力于为您提供更完美的热分析技术解决方案。继多对热电偶、非模型动力学分析、多频调制DSC、TGA-DSC同步分析、高频DMA技术及闪速FDSC之后,又为您推出新款用于常规材料分析的新型动态热机械分析仪DMA1。动态机械分析仪 (DMA) 用于测量材料机械性能及粘弹性能随温度、时间及频率的变化,新型DMA1操作方便,既可以进行传统DMA分析,而且可以使用静态力进行实验或者在液体中进行测试。灵活可旋转的的测试头使得测试不但可以在所有标准形变模式下进行,甚至可在液体或特定相对湿度条件下进行。DMA1是质量控制中既经济又可靠的理想选择。 注册参加网络研讨会后,您将获得有关这种技术的详细信息,并可以与梅特勒托利多的热分析应用技术专家直接讨论您的问题。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、参加及审核人数限制:限制报名人数为120人,审核人数100人。3、报名截止时间:2013年10月24日4、报名参会:http://simg.instrument.com.cn/meeting/images/20100414/baoming.jpg5、参与互动: *参会期间您还可以将有疑问的数据通过上传的形式给老师予以展示,并寻求解答*6、环境配置:只要您有电脑、外加一个耳麦就能参加。建议使用IE浏览器进入会场。7、提问时间:现在就可以在此帖提问啦,截至2013年10月23日8、会议进入:2013年10月24日13:30点就可以进入会议室9、特别说明:报名并通过审核将会收到1 封电子邮件通知函(您已注册培训课程),请注意查收,并按提示进入会议室!为了使您的报名申请顺利通过,请填写完整而正确的信息哦~http://simg.instrument.com.cn/webinar/20110223/images/zb_11.gif注意:由于参会名额有限,如您通过审核,请您珍惜宝贵的学习交流机会,按时参加会议。如您临时有事无法参会,请您进入报名页面请假。无故不参会将会影响您下一次的参会报名。快来参加吧:我要报名》》》
关于煤的水分的分析——水分测试 水是煤的不利杂质,水吸收煤燃烧产生的热,气化为水蒸气。煤中水分按结合状态可分为游离水和化合水两大类。游离水以吸附、附着等机械方式与煤结合;而化合水则以化合方式同煤中的矿物质结合,是矿物晶格的一部分,如硫酸钙(CaSO4·2H20)、高岭土(A12 03·2Si02·2H20)中的结晶水。煤的工业分析,只测定游离水。游离水按其赋存状态又分为外在水分和内在水分。煤的外在水分是指吸附在煤颗粒表面上或非毛细孔中的水分,在实际测定中是煤样达到空气干燥状态所失去的那部分水。煤的外在水分很容易蒸发,只要将煤放在空气中干燥,直到煤表面的水蒸气压和空气相对湿度平衡即可。煤的内在水分是指吸附或凝聚在煤颗粒内部毛细孔中的水。在实际测定中指煤样达到空气干燥状态时保留下来的那部分水。内在水在常温下不能失去,只有加热到一定温度时才能逸出。内在水分多少与煤的内表面积有关,内表面积越大,内在水分越高。不同变质程度煤的内表面积不同,变质程度愈浅,表面积愈大,其内在水分也愈高。 当煤颗粒中毛细孔吸附的水达到饱和状态时,此时的内在水分达到了最高值,称为煤的最高内在水分。最高内在水分在一定程度上能表示煤的煤化程度及某些煤质特征,可较好地区分低煤阶煤。至于因为开采、洗煤、运输、贮存等客观因素引人的水分,则是外在水分的主要来源。 由于煤粒大小不同,单位重量的表面积也不同。因此,和大气的接触面不同,则水分蒸发的难易也不同。为了使煤中的水分能迅速、1顶利地除去,必须将样品破碎至一定细度。但是,由于外在水分很容易失去。如果过度粉碎,水分就可能大量蒸发损失,结果造成测定数据远远不能表明煤中原有水分的实际含量。因此,为了测定数据能更接近实际情况,煤的工业分析规程规定的标准是先取小于13 mm的煤样,测定外在水分,然后,再粉碎至小于3mm测定内在水分。外在水分和经过换算的内在水分之和,称为应用基全水分(以符号叽表示)。但是,在生产实际中,也不一定全都分别测定,而可以在尽可能做到粉碎样品过程中,外在水分不致大量损失的条件下,将样品直接粉碎至3mm以下,然后,一次测定全水分(水分分析测试)。不论是经过上述哪种过程测定全水分,都因为样品粒度较大(3mm),水分不可能除尽。所以,为了精密测定及计算绝对干燥(或干燥基)样品的其他成分含量,还必须将风干样品粉碎至0.2mm以下,测定水分。测得的结果称为分析水分(以符号记表示)。因为测定条件不同,所以不能把分析水分解释为就是内在水分。通常分析水分必然稍高于内在水分。这些可在仪器仪表网中找寻水分仪测试来源——仪器仪表网
我是一个新手,问题如题,那位从事过均三嗪的液相分析,把条件共享一下吧,严重谢过!
【网络讲座】:安捷伦液相色谱技术在石油化工、煤化工分析中的最新应用【讲座时间】:2016年06月21日 10:00【主讲人】:左夏龙 安捷伦科技液相色谱应用工程师。化学、材料学专业背景。2013年加入安捷伦,从事液相色谱方法开发工作。【会议简介】本讲座以液相色谱常见的分离模式为脉络,结合相关标准,介绍液相色谱在石油化工和煤化工领域的典型应用。具体内容如下: 正相色谱测定中间馏分的芳香烃含量,反相色谱分析石化产品中的添加剂,以及体积排阻色谱分析石油化工下游的聚合物产品。 以反相色谱应用为例,介绍更快、更高分离度的超高效液相色谱技术。 基于阀切换技术的原油四组分全自动液相色谱分析方法以及多中心切割技术(MHC)在石化行业的应用介绍。 -------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名截止时间:2016年06月21日 9:304、报名参会:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/19195、报名及参会咨询:QQ群—171692483,扫码入群“色谱圈友荟”http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667772_2507958_3.gif
[color=#00FFFF][size=4][em0910][URL=http://www.hbhwkl.cn]http://www.hbhwkl.cn[/URL][/size][/color][煤的工业分析]煤的工业分析,又叫煤的技术分析或实用分析,是评价煤质的基本依据。在国家标准种,煤的工业分析包括煤的水分、灰分、挥发分和固定碳等指标的测定。通常煤的水分、灰分、挥发分和固定碳等指标的测定。通常煤的水分、灰分、挥发分是直接测出的,而固定碳是用差减法计算出来的。广义上讲,煤的工业分析还包括煤的全硫分和发热量的测定, 又叫煤的全工业分析。 1、煤的水分 煤的水分,是煤炭计价中的一个辅助指标。 煤的水分直接影响煤的使用、运输和储存。煤的水分增加,煤中有用成分相对减少,且水分在燃烧时变成蒸汽要吸热,因而降低了煤的发热量。煤的水分增加,还增加了无效运输,并给卸车带来了困难。特点是冬季寒冷地区,经常发生冻车,影响卸车,影响生产,影响车皮周转,加剧了运输的紧张。 煤的水分也容易引起煤炭粘仓而减小煤仓容量,甚至发生堵仓事故。 随着矿井开采深度的增加,采掘机械化的发展和井下安全生产的加强,以及喷露洒水、煤层注水、综合防尘等措施的实施,原煤水分呈增加的趋势。为此,煤矿除在开采设计上和开采过程中的采煤、掘进、通风和运输等各个环节上制定减少煤的水分的措施外,还应在煤的地面加工中采取措施减少煤的水分。 (1)煤中游离水和化合水 煤中水分按存在形态的不同分为两类,既游离水和化合水。游离水是以物理状态吸附在煤颗粒内部毛细管中和附着在煤颗粒表面的水分;化合水也叫结晶水,是以化合的方式同煤中矿物质结合的水。如硫酸钙(NaSO4.2H2O)和高龄土(AL2O3.2SiO2.2H2O) 中的结晶水。游离水在105~110C的温度下经过1~2小时可蒸发掉,而结晶水通常要在200C以上才能分解析出。 煤的工业分析中只测试游离水,不测结晶水。 (2)煤的外在水分和内在水分 煤的游离水分又分为外在水分和内在水分。 外在水分,是附着在煤颗粒表面的水分。外在水分很容易在常温下的干燥空气中蒸发,蒸发到煤颗粒表面的水蒸气压与空气的湿度平衡时就不再蒸发了。 内在水分,是吸附在煤颗粒内部毛细孔中的水分。内在水分需在100C以上的温度经过一定时间才能蒸发。 最高内在水分,当煤颗粒内部毛细孔内吸附的书分达到饱和状态时,这是煤的内在水分达到最高值,称为最高内在水分。最高内在水分与煤的孔隙度有关,而煤的孔隙度又于煤的煤化程度有关,所以,最高内在水分含量在相当程度上能表征煤的煤化程度,尤其能更好地区分低煤化度煤。如年轻褐煤的最高内在水分多在25%以上,少数的如云南弥勒褐煤最高内在水分达31%。最高内在水分小于2%的烟煤,几乎都是强粘性和高发热量的肥煤和主焦煤。无烟煤的最高内在水分比烟煤有有所下降,因为无烟煤的孔隙度比烟煤增加了。 (3)煤的全水分 全水分,是煤炭按灰分计加中的一个辅助指标。a.煤中全水分的含义。煤中全水分,是指煤中全部的游离水分,即煤中外在水分和内在水分之和。必须指出的是,化验室里测试煤的全水分时所测的煤的外在水分和内在水分,与上面讲的煤中不同结构状态下的外在水分和内在水分是完全不同的。化验室里所测的外在水分是指煤样在空气中并同空气湿度达到平衡时失去的水分(这是吸附在煤毛细孔中的内在水分也会相应失去一部分,其数量随当时空气湿度的降低和温度的升高而增大),这时残留在煤中的水分为内在水分。显然,化验室测试的外在水分和内在水分,除与煤中不同结构状态下的外在水分和内在水分有关外,还与测试是空气的湿度和温度有关。b.煤的全水分测试方法要点见GB212-91。 2、煤的灰分 煤的灰分,是指煤完全燃烧后剩下的残渣。因为这个残渣是煤中可燃物完全燃烧,煤中矿物质(除水分外所有的无机质)在煤完全燃烧过程中经过一系列分解、化合反应后的产物,所以确切地说,灰分应称为灰分产率
畜禽肉中己烯雌酚的测定测量不确定度分析依据JJF1059-1999《测量不确定度评定与表示》、CNAL/AG06《测量不确定度政策实施指南》和CNAL/AR11《测量不确定度政策》分析1目的对畜禽肉中己烯雌酚的测定的不确定度进行分析,找出影响不确定度的因素,对不确定度进行评估,如实反应测量的置信度和准确性。2适用范围畜禽肉中己烯雌酚的测定。3 职责3.1检测人员负责按操作规程操作仪器,确保测量过程中仪器正常运转,消除各种可能影响实验结果的意外因素,了解影响不确定度的因素。3.2校核人员负责检查原始记录及不确定度的计算方法。3.3技术人员负责审核检测结果和不确定度分析结果。4不确定度分析4.1. 测定方法按照国家标准GB/T5009.108-2003《畜禽肉中己烯雌酚的测定》中规定进行测定。 检测过程为:1) 称取样品:用精度为0.1mg的精密电子天平称取50.0000g样品,称准至0.1mg。2) 样品前处理:把样品置于300mL烧杯中,加入50mL水和100mL丙酮,用组织捣碎机提取1~2分钟。匀浆液过滤后,取滤液100ml于500mL分液漏斗中。向滤液中加入10-15g无水硫酸钠,充分摇动,静置分层;取上层有机相。下层水相用50mL二氯甲烷再提取一次。合并提取液,经装有20g-30g无水硫酸钠的玻璃漏斗脱水后滤入250ml茄型瓶中,再以40mL二氯甲烷分数次洗涤容器和无水硫酸钠,洗涤液也并入茄型瓶中,用旋转蒸发器浓缩后用二氯甲烷定容到10mL。装样上机,进样1.0uL。3) 己烯雌酚标准原液:使用国家标准物质中心提供的100mg/L的标准溶液。4) 己烯雌酚标准曲线溶液:将己烯雌酚标准原液用1ml移液管和100ml容量瓶稀释成1mg/L ;再用1ml移液管和100ml容量瓶把1mg/L的标准稀释成0.01mg/L;用5ml移液管和10ml容量瓶把1mg/L的标准稀释成0.5mg/L;用1ml移液管和10ml容量瓶把1mg/L的标准稀释成0.1mg/L;用1ml移液管和50ml容量瓶把1mg/L的标准稀释成0.05mg/L。共得到1mg/L、 0.5mg/L、0.1mg/L、0.05mg/mL、0.01mg/L的标准系列溶液。5) 样品的上机分析后,在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]上用5点校准曲线法进行定量。4.2. 测量流程方框示意图 4.3. 己烯雌酚含量的计算 式中:X——试样中己烯雌酚的含量,单位为毫克每千克(mg/kg);Aop——己烯雌酚的峰强度(峰面积);Aref——己烯雌酚标准的的峰强度(峰面积);Vop——最终定容体积,单位为毫升(mL);Vext——提取液总体积,单位为毫升(mL);Vclean——取用的提取液总体积,单位为毫升(mL);Msample——试样质量,单位为克(g);Rec——回收率。计算结果保留两位有效数字。 4.4 不确定度的来源本测试方法的不确定度来源主要来自如下几部分:4.4.1 测量重复性带来的不确定度分量 (含人员、环境、测量方法和仪器重复性等其他分量的贡献)。样品反复进行10次分析,见表1。表1 10.2050.00636×10-620.2110030.2040.00749×10-640.2300.019361×10-650.2240.013169×10-660.2130.0024×10-670.1990.011121×10-680.2130.0024×10-690.2010.010100×10-6100.2090.0024×10-6 0.211 (A类不确定度)4.4.2 样品测试全过程中引入的不确定度分量 。(B类不确定度)4.4.2.1 样品称取和前处理过程中天平准确度和容量瓶准确度引起的不确定度分量 。a 天平准确度引起的不确定度分量 天平的准确度为σ=0.1mg,采用矩型分布计算标准不确定度。 b 100ml容量瓶准确度引起的标准不确定度分量 。允差值为±0.1ml,按矩形分布。 C. 10ml容量瓶准确度引起的标准不确定度分量 允差值为±0.02ml,按矩形分布。 , 4.4.2.2标准溶液配制过程和GC/FPD制定校准曲线时引入的不确定度分量 。a). 己烯雌酚标准溶液引入的不确定度分量 。己烯雌酚标准溶液的浓度值为100mg/L±0.11mg/L,采用矩形分布。 b). 稀释过程中的移液管引起的不确定度分量 。1ml移液管的允差值为±0.008ml,5ml移液管的允差值为±0.025ml,都是矩形分布。1ml的移液管: , 5ml的移液管: , C). 容量瓶的准确度引起的不确定度分量 。10ml的容量瓶的允差值为±0.02ml,50ml的容量瓶的允差值为±0.05ml,100ml的容量瓶的允差值为±0.1ml,都是矩形分布。10ml的容量瓶: , 50ml的容量瓶: , 100ml的容量瓶: , d). 10μl进样针准确度引起的不确定度分量 。允差值为±0.02μl,矩形分布。 5. 合成标准不确定度计算标准不确定度一览表标准不确定度分量u(xi)不确定度来源标准相对不确定度 样品测量重复性0.0460 样品测试全过程( )0.0111 样品称取和前处理( )1.28×10-3 标准溶液配制过程和GC/FPD制定校准曲线( )0.0110 天平的准确度1.15×10-3 100ml容量瓶准确度5.77×10-4 10 ml容量瓶准确度1.15×10-3 己烯雌酚标准原液浓度5.77×10-4 标准稀释过程中移液管的准确度0.0107 标准稀释过程中容量瓶的准确度1.91×10-3 10μl进样针1.15×10-3合成相对不确定度: 6. 扩展不确定度的评定扩展不确定度通过使用包含因子k=2计算得出: 7.不确定度报告按照国家标准GB/T5009.108-2003规定的检测程序进行,最后测量出本次蔬菜水果中己烯雌酚含量为:0.21(2)mg/kg。[img]http://bbs.instrument.com.cn//images/affix.gif[/img][url=http://bbs.instrument.com.cn/download.asp?ID=194104]畜禽肉中己烯雌酚的测定测量不确定度分析.doc[/url] [color=#DC143C][size=4][font=黑体]应疯子哥的要求,把公式及图片贴上来,在9楼-13楼有版主ROGERSW的完整佳作。[/font][/size][/color]
请大家帮忙找找看有没有最新国外美洛昔康的红外光谱图,或者告诉我哪里能够找到,谢谢
一般来说,逻辑分析仪能看到比示波器更多的信号线。对于观察总线上的定时关系或数据 ——例如微处理器地址、数据或控制总线时,逻辑分析仪是特别有用的。逻辑分析仪能够解码微处理器的总线信息,并以有意义的形式显示。总之,当您通过了参数设计阶段,开始关注许多信号间的定时关系和需要在逻辑高和低电平码型上触发时,逻辑分析仪就是正确的测试工具。[b]逻辑分析仪[/b]大多数逻辑分析仪实际是合二而一的分析仪:一部分是定时分析仪,另一部分是状态分析仪。定时分析仪的信息显示形式与示波器的相同,水平轴代表时间,垂直轴代表电压幅度。由于这两种仪器上的波形都与时间相关,因此称为“时域”显示仪。[b]选择正确的采样方法[/b]定时分析仪好像是一台具有 1bit 垂直分辨率的数字示波器。由于只有 1bit 分辨率,因此只能实现两种状态 —高或低的显示。定时分析仪只关心用户定义的电压阈值。如果采样时信号高于该阈值,就以高或 1 显示,低于阈值的采样信号用低或0显示。从这些采样点得到一张由 1 和 0 组成,代表输入波形 1bit 图的表格。这张表格保存在存储器中,并可用来重建输入波形的 1bit 图,如图1所示。[align=center][url=http://www.elecfans.com/article/UploadPic/2008-11/200811278254695.jpg][img]http://www.elecfans.com/article/UploadPic/2008-11/200811278254695.jpg[/img][/url][/align][align=center][size=12px]图 1 定时分析仪的采样点[/size][/align]定时分析仪趋向于把各种信号拉成方波,这似乎会影响到它的可用性,但如果您需要同时观察几条甚至几百条信号线以验证信号间的定时关系,那么定时分析仪就是正确选择。应记住每个采样点都要使用一个存储器位置。分辨率越高(采样率越快),采集窗就越短。[b]跳变采样[/b]当我们捕获如图2 所示带有数据突发的输入线上的数据时,我们必须把采样率调到高分辨率(例如 4ns),以捕获开始处的快速脉冲。这意味着具有 4K(4096 样本)存储器的定时分析仪在 16.4ms 后将停止采集数据,使您不能捕获到第二个数据突发。[align=center][url=http://www.elecfans.com/article/UploadPic/2008-11/200811278255647.jpg][img]http://www.elecfans.com/article/UploadPic/2008-11/200811278255647.jpg[/img][/url][/align][align=center][size=12px]图2 高分辨率采样[/size][/align]在通常的调试工作中,我们采样和保存了长时间没有活动的数据。它们使用了逻辑分析仪存储器,却不能提供更多的信息。如果我们知道跳变何时产生,是正跳变还是负跳变,就能够解决这一问题。这一信息是有效使用存储器的跳变定时基础。为实现跳变定时,我们可在定时分析仪和计数器的输入处使用“跳变探测器”。现在定时分析仪只保存跳变前的那些样本,以及两个跳变之间的时间间隔。采用这种方法,每一跳变就只需使用两个存储器位置,输入无变动时就完全不占用存储器位置。在我们的例子中,根据每一突发中存在多少脉冲数,现在能捕获到第二、第三、第四和第五个突发。并同时保持达到 4ns 的高定时分辨率(图3)。[align=center][url=http://www.elecfans.com/article/UploadPic/2008-11/200811278255224.jpg][img]http://www.elecfans.com/article/UploadPic/2008-11/200811278255224.jpg[/img][/url][/align][align=center][size=12px]图3 使用跳变探测器采样[/size][/align][b]毛刺捕获[/b]毛刺脉冲因为会随机出现,造成灾难性的后果而声名狼藉。定时分析仪可采样输入数据,保持对采样间所产生任何跳变的跟踪,容易捕获毛刺。在分析仪中,把毛刺定义为相邻两次采样间穿越逻辑阈值一次以上的任何跳变。为了识别毛刺,我们要“教会”分析仪保持对所有多个异常跳变的跟踪,并将它们作为毛刺显示。毛刺显示是一种很有用的功能,能够提供毛刺触发和显示超前毛刺的数据,从而帮助我们确定毛刺产生的原因。这种能力也使得分析仪只捕获毛刺产生时所要的数据。回顾本节开始时提到的例子。我们有一个系统周期性地因毛刺出现在一条信号线上而崩溃。由于毛刺发生具有偶然性,您即使能保存整个时间上所有数据(假定有足够的存储能力),也很难在巨大的信息量中找到它。另一种方法是使用没有毛刺触发功能的分析仪,您必须坐在仪器前,按运行按钮,等待看到毛刺为止。[b]定时分析仪的触发[/b]逻辑分析仪连续捕获数据,并在找到跟踪点后停止采集。这样,逻辑分析仪就能显示出被称为负时间的跟踪点前的信息,以及跟踪点后的信息。[b]码型触发[/b]设置定时分析仪的跟踪特性与设置示波器的触发电平和斜率稍有一点区别。许多分析仪是在跨多条输入线的高和低码型上触发。为使某些用户更感方便,绝大多数分析仪的触发点不仅可用二进制( 1 和 0),而且可用十六进制、八进制、ASCII或十进制设置。在查看4、 8、16、24、32bit宽的总线时,使用十六进制的触发点会更加方便。设想如果用二进制设置24bit总线就会麻烦得多。[b]边沿触发[/b]在调节示波器的触发电平旋钮时,您知道是在设置电压比较器的电平,这个电平将告诉示波器在输入电压穿越该电平时触发。定时分析仪的边沿触发与其基本相似,但触发电平已预设置到逻辑阈值。大部分逻辑器件都与电平相关,这些器件的时钟和控制信号通常都对边沿敏感。边沿触发使您能与器件时钟同步地捕获数据。您能告诉分析仪在时钟边沿产生(上升或下降)时捕获数据,并获取移位寄存器的所有输出。当然在这种情况下,必须延迟跟踪点,以顾及通过移位寄存器的传播延迟。[b]状态分析仪基础[/b]如果您从未使用过状态分析仪,您可能认为这是一种极为复杂的仪器,需要花很多时间才能掌握使用方法。事实上,许多硬件设计师发现状态分析仪中有许多极有价值的工具。一个逻辑电路的“状态”是数据有效时对总线或信号线的采样样本。例如,取一个简单的“D”触发器。“D”输入端的数据直到时钟正沿到来时才有效。这样,触发器的状态就是正时钟沿产生时的状态。现在,假定我们有8个这样的触发器并联。所有8个触发器都连到同样的时钟信号上。当时钟线上产生正跳变时,所有8个触发器都要捕获各自“D”输入的数据。这样,每当时钟线上正跳变时就产生一个状态,这8条线类似于微处理器总线。如果我们把状态分析仪接到这8条线上,并告诉它在时钟线正跳变时收集数据,状态分析仪将照此执行。除非时钟跳到高电平,否则输入的任何活动将不被状态分析仪捕获。定时分析仪由内部时钟控制采样,因此它是对被测系统作异步采样。而状态分析仪从系统得到采样时钟,因此它是对系统同步采样。状态分析仪通常用列表方式显示数据,而定时分析仪用波形图显示数据。[b]理解时钟[/b]在定时分析仪中,采样是沿着单一内部时钟的方向进行,从而使事情非常简单。但微处理器系统中往往会有若干个“时钟”。假定某个时刻我们要在RAM中的一个特定地址上触发,并查看所保存的数据;再假定使用的微处理器是Zilog公司的 Z80。为了用状态分析仪从Z80捕获地址,我们要在MREQ线为低时进行捕获。而为了捕获数据,需要在WR线为低(写周期)或RD线为低(读周期)时让分析仪采样。某些微处理器可在同一条线上对数据和地址进行多路转换。分析仪必须能让时钟信息来自相同的信号线,而非来自不同的时钟线。[align=center][url=http://www.elecfans.com/article/UploadPic/2008-11/200811278255919.jpg][img]http://www.elecfans.com/article/UploadPic/2008-11/200811278255919.jpg[/img][/url][/align][align=center][size=12px]图 4 RAM 定时波形图[/size][/align]在读写周期期间,Z80首先把一个地址放在地址总线上。接着设定MREQ线在该地址对存储器的读或写有效。最后根据现在是读还是写对RD或WR线断言。WR线只有在总线数据有效后才被设定。这样,定时分析仪就作为多路分配器在适当的时间捕获地址,然后在同一信号线上捕获产生的数据。[b]触发状态分析 [/b]像定时分析仪一样,状态分析仪也提供限定所要保存数据的功能。如果我们要寻找地址总线上由高低电平构成的特定码型,可告诉分析仪在找到该模式时开始保存,直到分析仪的存储器完全装满。这些信息可以用十六进制或二进制格式显示。但在解码至汇编码时,十六进制可能更为方便。在使用处理器时,应把这些特定的十六进制字符与处理器指令相比较。大多数分析仪制造商设计了称为反汇编器的软件包,这些软件包把十六进制代码翻译成易于阅读的汇编码。[align=center][url=http://www.elecfans.com/article/UploadPic/2008-11/200811278255303.jpg][img]http://www.elecfans.com/article/UploadPic/2008-11/200811278255303.jpg[/img][/url][/align][align=center][size=12px]图 5 把十六进制码翻译成汇编码[/size][/align][b]序列级和选择性保存[/b]状态分析仪具有帮助触发和存储的“序列级”数据。序列级使您能比单一触发点更精确地限定要保存的数据。也就是说可使用更精确的数据窗,而不必存储不需要的信息。选择性的保存意味着可只保存较大整体中的一部分。例如,假定我们有一个计算给定数平方的汇编例程。如果该例程不能正确计算平方,我们就告诉状态分析仪捕获这一例程。具体做法是先让状态分析仪寻找该例程的起点。当它找到起始地址时,我们再告诉它寻找终止地址,并保存两者之间的所有信息。当发现例程结束时,我们告诉分析仪停止状态保存。[b]探测解决方案[/b]为进行调试,向数字系统施加的物理连接必须方便可靠,对被调试的目标系统只有最小的侵扰,这样才能使逻辑分析仪得到精确的数据。普通的探测解决方案是每条电缆有 16 个通道的无源探头。每个通道的两端用100kΩ并联8pF 端接。您可将这种无源探头与示波器探头的电气性能作一比较。无源探测系统除了更小的尺寸和更高的可靠性外,还能把探头端接在与目标系统的连接点上。这就避免了从大的有源探头接口夹到被测电路之间大量引线所产生的附加杂散电容。因此您的被测电路就只“看到”8pF的负载电容,而不再是前述探测系统的16pF。[align=center][url=http://www.elecfans.com/article/UploadPic/2008-11/200811278255595.jpg][img]http://www.elecfans.com/article/UploadPic/2008-11/200811278255595.jpg[/img][/url][/align][align=center][size=12px]图6 分析探头[/size][/align]把状态分析仪接到微处理器系统需要进行机械连接和时钟选择。某些微处理器可能需要外部电路对一些信号进行解码,才能得到用于状态分析仪的时钟。分析探头不仅能提供与目标系统快速、可靠和正确的机械连接,而且能提供必要的电气适配能力,如为正确捕获系统运行提供的时钟和多路分配器。[b]结语[/b]绝大多数逻辑分析仪都由定时分析仪和状态分析仪这两个主要部分组成。定时分析仪更适于处理多线的总线型结构或应用。它能够在信号线上的码型上,甚至在毛刺上触发。状态分析仪常被看成是一种软件工具,事实上它在硬件设定也很有用。由于它从被测系统得到时钟,因此捕获的数据也就是系统在时钟上的数据。逻辑分析仪为数字电路设计工程师提供了强大的设计工具。[table=349][tr][td][url=https://yqj.mumuxili.com/?from=YQSQ2-7/1]https://yqj.mumuxili.com/?from=YQSQ2-7/2[/url][/td][/tr][/table]
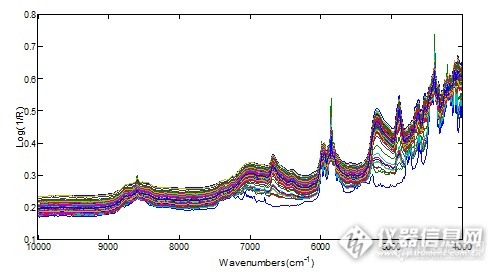
[align=center][b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术用于美洛西林钠舒巴坦钠药物混合过程在线混合均匀度终点监测[/b][/align][align=left][b]摘要: [/b]利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术,对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。[/align][b]关键词[/b]:[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url];分析模型;混合均匀度;在线监测自从2004年美国食品与药品监督管理局提出“过程分析技术”以来,全球的药品生产企业正在向着更高技术含量的生产方式和质量控制方式进军。近红外(Near infrared,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])光谱分析技术因其快速,无损的特点成为“过程分析技术”的重要组成部分,是制药企业进行产品中间体质量控制的重要方法之一。传统的检测方法为高效液相色谱法,紫外可见分光光度法等需要停止混合操作时才能取样检测,并且等待检测结果所需的时间也比较长,工作效率比较低,而[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱可以进行在线检测,连续记录不同混合时间内混合物的光谱图,建立数学模型对采集数据进行分析,从而判断各组分之间是否已经达到质量均一,工作效率大幅度的提高。本研究利用 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] 光谱分析技术在线监测美洛西林钠舒巴坦钠的药物混合过程,从而实现混合终点的准确判断。[b]1 材料1.1试剂[/b]美洛西林钠(13102041,山东瑞阳制药有限公司)舒巴坦钠(SS201310-26,江西东风制药有限公司)[b]1.2仪器和软件[/b]AntarisII型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国ThermoFisher公司),附有积分球采样模块;RESULT采样软件;电子分析天平(Sartorius BT224S,德国);TQ数据处理软件;表面皿;药匙;自制搅拌器。[b]2 方法2.1样品的准备[/b]精密称取舒巴坦钠固体原料药10.00g,美洛西林钠固体原料药40.00g,以备进行在线混合光谱的采集。平行制备3批样品,进行混合光谱的采集。[b]2.2模型的建立[/b]目前,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于混合过程在线监测的方法可分为活性药物成分(API)定量分析模型监测和基于移动块标准偏差(MBSD)的定性分析模型监测。前者为基于API药物含量的定量监测模型,当达到混合终点时,API的含量趋于一定值,可以依据模型监测的含量是否达到理论值并趋于稳定进行混合终点的监测;后者为基于光谱的标准偏差的定性监测模型。MBSD法的基本原理为:连续采集的若干张光谱间的标准偏差变化率趋于稳定并小于限定的一阈值时可认为达到了混合终点。其具体的计算步骤为:首先确定用于计算光谱标准偏差的光谱的条数n(即移动块的宽度),当[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析仪器采集到n张光谱后计算n张光谱的峰面积(或最大峰高、平均峰高等)的标准差,当采集到n+1张光谱时将第一张光谱移除,计算最近n张光谱的标准差,如此类推,最终得到随时间变化的光谱的标准偏差,根据标准差的变化进行混合终点的监测。本研究中建立了舒巴坦钠含量的定量分析模型和基于MBSD法的定性分析模型同时对用于混合终点的判断。[b]2.3在线混合光谱的采集[/b]将称取的美洛西林钠、舒巴坦钠原料药样品放入表面皿中,然后将表面皿放在Antaris II型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]积分球采样模块的上面,采用积分球漫反射采样方式进行光谱的采集。在运行在线混合工作流的同时采用自制的搅拌器进行样品的混合,采集得到混合过程的原始光谱,同时监测混合过程。波长范围10000-4000cm[sup]-1[/sup],每张光谱扫描次数4,混合过程中每间隔5s进行一张光谱的采集,光谱分辨率为8.0cm[sup]-1[/sup],每4个小时进行背景光谱的采集。每张[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱由1557个变量点组成。[b]2.4定量定性分析模型用于终点判断数据分析[/b]将在线混合过程进行监测,得到在线混合过程数据进行分析,以便了解混合全过程信息以及混合过程的监测。[b]2.5混合终点分析[/b]当得到混合终点时分别采集混合后的样品6处的原始[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,利用舒巴坦钠的定量分析模型预测混合终点时不同样品点处的舒巴坦钠的含量,判别是否混合均匀。[b]3 实验结果3.1分析模型的建立[/b]本研究中分别建立了在线混合过程的舒巴坦钠定量监测模型和基于移动块标准偏差的定性监测模型。[b]3.1.1 定性分析模型的建立[/b]目前混合均匀度在线监测常用的方法为MBSD法,本研究中MBSD法定性建模的参数为:选择的3个光谱区间包括全光谱、5275.6-4806.3cm[sup]-1[/sup](称为Region1)及7096.76-6344.66cm[sup]-1[/sup](称为Region2);用于计算光谱偏差的光谱的条数为5(即移动块的宽度为5)。[b]3.1.2 定量分析模型的建立[/b]本研究中所建立的定量分析模型用于监测混合过程中舒巴坦钠的百分含量的变化,因为本实验中舒巴坦钠和美洛西林钠两者间的混合比为4:1,当达到混合终点时,舒巴坦钠的百分含量应该在20%左右。其模型的具体参数见上一章中得到的舒巴坦钠百分含量的定量分析模型。[b]3.2混合在线过程数据分析[/b]本研究中平行进行了3次混合过程的在线监测,分别对3次实验结果进行分析,以充分了解混合监测过程。[b]3.2.1 第一批实验结果分析3.2.1.1 原始光谱图[/b]图1给出了混合过程中采集得到的208张原始光谱,由图中可知,处于下面的光谱较稀疏,可能属于混合刚开始的阶段,光谱会有较大的差异;处于上面的光谱较密集,其原因为随着混合的不断进行,光谱间差异越来越小,所以光谱较集中。[align=center][img=,498,274]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141912_01_1626619_3.png[/img][/align][align=center]图1 第一批混合过程原始光谱[/align][align=center] [/align][b]3.2.1.2 在线混合过程结果分析[/b]图2为定性分析模型中得到的3个光谱区间的峰面图,其中M1为全光谱建模的峰面积变化,M2为Region 1(5275.6-4806.3cm-1)的峰面积变化,M2为Region 2(7096.76-6344.66cm-1)的峰面积变化,由峰面积的变化图可知,混合过程的前100s其变化较为明显,M1不断升高,M2和M3(7096.76-6344.66cm-1)不断下降,之后峰面积值趋于稳定。[align=center][img=,525,234]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141913_01_1626619_3.png[/img][/align][align=center]图2 光谱区间峰面积图[/align]图3为舒巴坦钠含量及标准偏差变化图,由图中显示在混合的初期阶段,尤其是前100s左右,四个表征混合均匀度的参数均有着较大的变化趋势,在200-300s间四个参数有稍微较小的波动,此后随着混合过程的不断进行,表征混合均匀度的四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右,舒巴坦钠和美洛西林钠混合较为均匀,达到了混合终点。由图可知前100s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,538,292]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141914_01_1626619_3.png[/img][/align][align=center]图 3 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align][align=left] 当达到混合终点时分别采集表面皿下6个点的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,根据建立的模型测定其舒巴坦钠的百分含量,看混合是否均匀。表2给出了用所建模型得到的6个点的舒巴坦钠的百分含量值,6个点舒巴坦钠的百分含量值在20%左右,说明混合较为均一,但是最大的值达到了22.41%,可能是由于混合装置过于简陋,加上是人为搅拌进行混合,不能达到很好的混合,部分地方没有进行很好的混合。从实验的可行性方面,初步证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]技术用于美洛西林钠舒巴坦钠混合的可行性。[/align][align=center]表1混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,570,70]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_01_1626619_3.png[/img][/align][b]3.2.2 第二批实验结果分析3.2.2.1 原始光谱图[/b]图4给出了第二批混合过程中采集得到的203张原始光谱,其混合过程原始光谱的特征和第一批混合过程较为相似,混合初期光谱变化较为明显,随着混合的进行,光谱差异变小,光谱较为密集。[align=center][img=,488,280]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_02_1626619_3.png[/img][/align][align=center]图4 第二批混合过程原始光谱[/align][align=left] [b]3.2.2.2 在线混合过程结果分析[/b][/align]图5为各个光谱波段峰面积的变化图,由图中显示开始的100s内峰面积有着较大的变化幅度,随着混合的不断进行,峰面积的变化趋势不断减小并逐渐趋于稳定。[align=center][img=,516,307]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141916_01_1626619_3.png[/img][/align][align=center]图5 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图6为舒巴坦钠含量及标准偏差变化图,由图可知在混合的初期阶段大约0-100 s时,舒巴坦钠百分含量值及峰面积的标准偏差值有着明显的变化,全光谱峰面积的标准偏差(Full Range STD)在200-400 s间有较为明显的波段,此后随着混合过程的不断进行,四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右。由此可知前100 s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,551,327]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141917_01_1626619_3.png[/img][/align][align=center]图6 含量和标准偏差变化图[/align][align=center](a 舒巴坦钠百分含量 b 全光谱峰面积标准偏差 c Region 1峰面积标准偏差 d Region 2峰面积标准偏差)[/align]当达到混合终点时,采集表面皿底部6处的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,检测混合过程是否达到均一,表2列出来了6处的舒巴坦钠的百分含量值,由表2可知达到混合结束后得到的6处的舒巴坦钠的百分含量均在20%左右,说明混合较为均匀。同时,由于实验条件的限制加上搅拌时人为因素的影响等,各点之间含量也着较大的差异。[align=center]表2 舒巴坦钠百分含量[/align][align=center] [img=,566,84]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141918_01_1626619_3.png[/img][/align][b]3.2.3 第三批实验结果分析3.2.3.1 原始光谱图[/b]图7给出了混合过程中采集得到的207张原始光谱,由图中可知,得到的原始光谱图与第一批和第二批有着相似的结果,即混合的初期光谱差异大,因此光谱较为稀疏(偏下方的光谱),随着混合的进行,光谱间差异变小,光谱变得密集(偏上方的光谱)。[align=center][img=,505,262]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_01_1626619_3.png[/img][/align][align=center]图7 第三批混合过程原始光谱[/align][b]3.2.3.2 在线混合过程结果分析[/b]图8给出了混合过程中3个光谱区间峰面积的变化趋势值,由图中可知0-100s间三个光谱区间的峰面积有着明显的变化,100-200s间峰面积有着明显的变化,但是变化幅度没有前100s大,200s以后峰面积变化趋势变小。说明前200s是混合的主要阶段,峰面积变化较为明显。[align=center][img=,519,343]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_02_1626619_3.png[/img][/align][align=center]图 8 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图9为舒巴坦钠百分含量及光谱峰面积的标准偏差随时间变化的趋势图,其变化趋势和峰面积的变化趋势相似,前100s变化幅度较大,100-200s间也有较为明显的变化,但是变化幅度不是很明显,200s后舒巴坦钠的百分含量和峰面积的标准偏差均趋于稳定,说明此时光谱差异变小,混合趋于均匀。[align=center][img=,529,352]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141920_01_1626619_3.png[/img][/align][align=center]图9 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align]表3为达到混合终点时采集表面皿底部的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱得到的不同点的舒巴坦钠的百分含量值,由表中显示6个点的舒巴坦钠的百分含量值在20%左右,但是6个点之间舒巴坦钠百分含量间存在较大的差异,测得的最小值为17.80%,其原因可能是一方面由于实验条件的限制混合不够均匀,一方面用于舒巴坦钠含量测定的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]定量分析模型也有一定的偏差,可能引起含量检测的差异存在。[align=center]表3 混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,564,66]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141921_01_1626619_3.png[/img][/align][b]3.3小结[/b]通过3个混合平行实验的进行可知所建立的基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型能够有效的监测舒巴坦钠、美洛西林钠的混合过程。由舒巴坦钠百分含量和标准偏差变化图可知两者的变化有着相关性,当舒巴坦钠的百分含量变化幅度大时,其标准偏差的变化幅度也较大,因此两者均可以用于混合过程的在线监测,证实了实验的可行性。[b]4 结论和讨论[/b]本研究采用AntarisII傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,然后Antaris II傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]漫反射采样方式采集混合过程中的光谱,实时监测混合过程的进行。通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。此外,MBSD法因为无需进行一级数据的采集,方法较为简单且容易理解,目前常用于混合过程的在线监测。本研究中有效证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术在舒巴坦钠美洛西林钠样品在线混合过程中应用的可行性,在样品的在线混合监测中有着重要的应用价值和应用前景。该技术能够克服传统方法费时、繁琐等缺点,而且可以实现过程的实时在线监测,让生产者充分了解整个生产过程中的参数变化。 [b]参考文献[/b]陆婉珍, 褚小立. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]([url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])和过程分析技术(PAT). 现代科学仪器, 2007(004):13-17.SieslerH, Ozaki Y, Kawata S, et al. Near-infrared spectroscopy: principles .Instruments, Applications, 2002:35-181.Bhushan,K.R.,et al.Detection of breastcancer microcalcifications using a dual-modality SPECT/[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] fluorescent probe. J Am Chem Soc, 2008. 130(52):17648-17649.贾燕花. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术在化学药品生产过程控制应用初探. 北京协和医学院, 2011.Fevotte.G,et al.Applications of [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]spectroscopy to monitoring and analyzing the solid state during industrialcrystallization processes . Int J Pharm, 2004, 273(1):159-169.张敏.盐酸林可霉素多晶型分子构象对其红外光谱行为的影响.中国抗生素杂志, 2005, 30(009):529-532.Blanco M,R Goz"01ez Ba,E.Bertran,Monitoring powder blending in pharmaceutical processes by use of nearinfrared spectroscopy . Talanta, 2002, 56(1):203-212,田科雄.不同装载系数和混合时间对添加剂预混料混合均匀度的影响.河北畜牧兽医, 2004, 20(9):52-53.孙栋. 基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的几种固体粉末混合均匀度快速检测研究. 山东大学硕士学位论文, 2012年.
仪器仪表网介绍关于煤的水分的分析——水分测试 水是煤的不利杂质,水吸收煤燃烧产生的热,气化为水蒸气。煤中水分按结合状态可分为游离水和化合水两大类。游离水以吸附、附着等机械方式与煤结合;而化合水则以化合方式同煤中的矿物质结合,是矿物晶格的一部分,如硫酸钙(CaSO4·2H20)、高岭土(A12 03·2Si02·2H20)中的结晶水。煤的工业分析,只测定游离水。游离水按其赋存状态又分为外在水分和内在水分。煤的外在水分是指吸附在煤颗粒表面上或非毛细孔中的水分,在实际测定中是煤样达到空气干燥状态所失去的那部分水。煤的外在水分很容易蒸发,只要将煤放在空气中干燥,直到煤表面的水蒸气压和空气相对湿度平衡即可。煤的内在水分是指吸附或凝聚在煤颗粒内部毛细孔中的水。在实际测定中指煤样达到空气干燥状态时保留下来的那部分水。内在水在常温下不能失去,只有加热到一定温度时才能逸出。内在水分多少与煤的内表面积有关,内表面积越大,内在水分越高。不同变质程度煤的内表面积不同,变质程度愈浅,表面积愈大,其内在水分也愈高。 当煤颗粒中毛细孔吸附的水达到饱和状态时,此时的内在水分达到了最高值,称为煤的最高内在水分。最高内在水分在一定程度上能表示煤的煤化程度及某些煤质特征,可较好地区分低煤阶煤。至于因为开采、洗煤、运输、贮存等客观因素引人的水分,则是外在水分的主要来源。 由于煤粒大小不同,单位重量的表面积也不同。因此,和大气的接触面不同,则水分蒸发的难易也不同。为了使煤中的水分能迅速、1顶利地除去,必须将样品破碎至一定细度。但是,由于外在水分很容易失去。如果过度粉碎,水分就可能大量蒸发损失,结果造成测定数据远远不能表明煤中原有水分的实际含量。因此,为了测定数据能更接近实际情况,煤的工业分析规程规定的标准是先取小于13 mm的煤[fo
我国是以煤炭为主要一次能源的国家,一次能源消费中煤炭的占比达到62%。但我国的煤炭利用技术总体上是落后的,在煤炭的转化利用过程中普遍存在效率低、污染严重等问题。随着能源问题的日益突出,洁净煤技术越来越多地应用于实际生产过程中,其中大规模煤气化、煤气化多联产技术成为了煤炭综合应用的主要方向之一。“十一五”期间,煤气化属于国家鼓励项目,其中明确指出新型煤化工领域将重点开发和实施煤的焦化技术、大型煤气化技术和以煤气化为核心的“多联产”技术。2. 煤气化原理煤炭气化是指煤在特定的设备内,在一定温度及压力下使煤中有机质与气化剂(如蒸汽/空气或氧气等)发生一系列化学反应,将固体煤转化为含有CO、H2、CH4等可燃气体和CO2、N2等非可燃气体的过程。气化过程发生的反应包括煤的热解、气化和燃烧反应。煤的热解是指煤从固相变为气、固、液三相产物的过程。煤的气化和燃烧反应则包括两种反应类型,即非均相气-固反应和均相的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]反应。煤炭气化时,必须具备三个条件,即气化炉、气化剂、供给热量,三者缺一不可。煤气化工艺根据气化炉内煤料与气化剂的接触方式不同可区分为固定床(移动床)、流化床、气流床,此外还有地下煤气化工艺。3. 煤气分析仪的原理和技术特点近年来红外煤气分析仪越来越多地应用于实际煤气化煤气分析当中。 红外煤气分析仪采用红外传感器测量煤气成分中的CO、CO2、CH4、CnHm的浓度,使用热导传感器测量H2的浓度,使用电化学传感器测量O2浓度,同时根据测量成分的浓度,计算得到煤气的理论热值。红外煤气分析仪取代了奥氏气体分析仪的人工取样和人工分析环节,可实现自动化测量,避免了人工误差;同时预处理系统和仪器相对燃烧法热值仪具有结构简单,操作维护方便的特点,更加适合煤气化实时在线的分析要求。红外煤气分析仪具备H2测量补偿功能,保证了H2浓度的准确测量。热导传感器用于测量多种混合气体时,必然要考虑到煤气中其他气体的影响因素。煤气主要成分中CO、O2 与背景气N2的热导系数相当,对H2的测量结果影响不大,但是CO2 、CH4 对H2测量影响明显。通过理论分析及实验表明,如果气体成分中含有CO2,会使H2的测量读数偏低;如果气体成分中含有CH4,会使H2的测量读数偏高。因此为了得到准确的H2含量,应对H2浓度进行CO2 、CH4的浓度校正。煤气分析仪对煤气的各气体成分进行分析,并将各种气体的相互影响进行了浓度修正和补偿,消除煤气中其他成分对H2的影响,保证了H2测量值的准确性。此外 煤气分析仪采用了旁流扩散式的热导检测池,流量在0.3―1.5L/min的范围内变化对热导的测量没有影响,减少了因流量波动造成H2测量的误差影响。煤气化过程中产生的煤气中的碳氢化合物除了CH4外,还有少量的CnHm,大多数红外分析仪仅以CH4为测试对象,折合成碳氢化合物总量计算热值。根据红外吸收原理,如图1,乙烷等碳氢化合物在甲烷的特征波长3.3um左右有明显吸收干扰。当煤气中其他碳氢化合物含量较大时,CH4的测试值会明显偏大,导致热值测试不准,其热值测试值也无法保证精度。甲烷、乙烷、丙烷、丁烷的红外吸收光谱图1:甲烷、乙烷、丙烷、丁烷的红外吸收光谱红外煤气分析仪采用了特殊的气体滤波技术,可实现无干扰的CH4测量,准确反应混合煤气中CH4和CnHm成分的实际变化,有利于热值的准确分析。4. 煤气分析仪在煤气化中的应用根据煤气化应用领域的不同,煤气分析仪可实现煤气热值分析和煤气成分分析两种用途。通常的应用如下:4.1 工业燃气应用作为工业燃气,一般热值要求为1100-1350大卡热的煤气,可采用常压固定床气化炉、流化床气化炉均可制得。主要用于钢铁、机械、卫生、建材、轻纺、食品等部门,用以加热各种炉、窑,或直接加热产品或半成品。实际应用中通常需要控制加热温度,以达到工艺或质量控制目的,燃气的热值稳定性就尤为重要。红外煤气分析仪针对H2和CH4的测量采用了测量补偿技术,可保证实际热值测试结果的准确性,为燃气的燃烧测控提供了有效有力的数据依据。4.2 民用煤气应用民用煤气的热值一般在3000-3500大卡,同时还要求CO小于10%,除焦炉煤气外,用直接气化也可得到,采用鲁奇炉较为适用。与直接燃煤相比,民用煤气不仅可以明显提高用煤效率和减轻环境污染,而且能够极大地方便人民生活,具有良好的社会效益与环境效益。出于安全、环保及经济等因素的考虑,要求民用煤气中的H2、CH4、及其它烃类可燃气体含量应尽量高,以提高煤气的热值;而CO有毒其含量应尽量低。 红外煤气分析仪测试煤气热值可知道气化站的煤气混合,保证燃气热值;同时可测得CO、H2、CH4的实际浓度,有效控制CO浓度,保证燃气安全。4.3 冶金还原气应用煤气中的CO和H2具有很强的还原作用。在冶金工业中,利用还原气可直接将铁矿石还原成海棉铁;在有色金属工业中,镍、铜、钨、镁等金属氧化物也可用还原气来冶炼。因此,冶金还原气对煤气中的CO含量有要求。 红外煤气分析仪可实时有效测量CO或H2浓度,指导调整气化工艺,保证产气效率。4.4 化工合成原料气随着新型煤化工产业的发展,以煤气化制取合成气,进而直接合成各种化学品的路线已经成为现代煤化工的基础,主要包括合成氨、合成甲烷、合成甲醇、醋酐、二甲醚等。化工合成气对热值要求不高,主要对煤气中的CO、H2等成分有要求,一般德士古气化炉、Shell气化炉较为合适。目前我国合成氨的甲醇产量的50%以上来自煤炭气化合成工艺。若煤气成分中CO2浓度过高,直接会影响合成工序压缩机的运行效率(一般降低10%左右),必然造成电耗和压缩机维修费用增加。红外煤气分析仪用于CO、CO2、H2等气体的浓度测量,用于指导合成气工艺控制,可保证化工产品的产量和质量,同时可达到节能的目的。4.5 煤制氢应用氢气广泛的用于电子、冶金、玻璃生产、化工合成、航空航天、煤炭直接液化及氢能电池等领域,目前世界上96%的氢气来源于化石燃料转化。而煤炭气化制氢起着很重要的作用,一般是将煤炭转化成CO和H2,然后通过变换反应将CO转换成H2和H2O,将富氢气体经过低温分离或变压吸附及膜分离技术,即可获得氢气。实际应用中由于CO含量的增加,必然会导致变换工序中变换炉的负荷增加。它不但会使催化剂的使用寿命缩短,而且使变换炉蒸汽消耗增加。红外煤气分析仪用于煤气成分分析,提供煤气中各气体成分的浓度数据,指导气化和转换工艺的控制,可起到节能增效的作用。此外, 红外煤气分析仪还可在煤气化多联产的应用中提高化工生产效率,提供清洁能源,改进工艺过程,以达到效益大化,有助于提升产业技术水平。5. 结论随着煤气化技术在国内的应用和发展,对于煤气化过程的监测和控制提出了更高的要求。 红外煤气分析仪集成了红外、热导和电化学三种气体传感器技术,可实现对煤气的成分分析和热值分析。在实际应用中解决了H2测量补偿和CH4测量抗干扰的问题,更广泛地应用于工业燃气、民用煤气、冶金、化工等行业,可指导工艺控制和改善,并达到节能增效的作用,有利于促进煤气化技术的提升。
[align=center][img]https://img1.17img.cn/17img/images/202103/webinar/36af542f-66e9-44ec-897d-7a91fa340fc4.jpg[/img][/align]颗粒学研究包罗万象,涉及食品、医药、化工、材料、冶金等各行各业。2020年,席卷全球的新型冠状病毒平均直径约为100纳米,属于纳米颗粒,新冠病毒的气溶胶传播也属于颗粒研究的范畴。疫情进一步推动颗粒学的研究与应用向着更小、更复杂、更尖端的纵深快速发展,同时,颗粒研发与质控所必须的相关检测分析技术也在不断迭代升级。基于此,仪器信息网联合中国颗粒学会,将于2021年3月24日-3月26日组织召开第二届“颗粒研究应用与检测分析”主题网络会议。分设[b]能源颗粒和电池材料、药物制剂与粒子设计、气溶胶与新冠病毒、超微及纳米颗粒、颗粒测试与表征[/b]五个分会场,邀请业内著名颗粒学学者、检测分析专家及企业代表,针对颗粒学研究应用及检测分析的前沿热点和疑难问题进行探讨,为颗粒学的研发应用端与检测分析端搭建交流平台。热忱欢迎国内外颗粒领域的专家、学者、技术人员、企业界代表及研究生踊跃参会、交流。报名链接:https://insevent.instrument.com.cn/t/w2
【网络讲堂】:智能化过程分析优化研发、放大和生产【讲座时间】:2015年02月05日 14:30【主讲人】:胡建斌 (梅特勒-托利多(上海)过程检测部培训经理,在梅特勒公司14年工作经验,熟悉过程分析的产品和应用,了解行业未来的发展方向。)【会议简介】智能化是过程分析的发展方向,不断的创新使得在线仪表不仅能提供常规的测量参数,例如pH、溶解氧等,更能够告诉用户什么时候需要对探头进行校准,什么时候应该更换探头等智能化的诊断信息。-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2015年02月05日 14:004、报名参会:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/13335、报名及参会咨询:QQ群—231246773
煤的工业分析煤的工业分析,又叫煤的技术分析或实用分析,是评价煤质的基本依据。在国家标准种,煤的工业分析包括煤的水分、灰分、挥发分和固定碳等指标的测定。通常煤的水分、灰分、挥发分和固定碳等指标的测定。通常煤的水分、灰分、挥发分是直接测出的,而固定碳是用差减法计算出来的。广义上讲,煤的工业分析还包括煤的全硫分和发热量的测定, 又叫煤的全工业分析。1、煤的水分 煤的水分,是煤炭计价中的一个辅助指标。 煤的水分直接影响煤的使用、运输和储存。煤的水分增加,煤中有用成分相对减少,且水分在燃烧时变成蒸汽要吸热,因而降低了煤的发热量。煤的水分增加,还增加了无效运输,并给卸车带来了困难。特点是冬季寒冷地区,经常发生冻车,影响卸车,影响生产,影响车皮周转,加剧了运输的紧张。 煤的水分也容易引起煤炭粘仓而减小煤仓容量,甚至发生堵仓事故。 随着矿井开采深度的增加,采掘机械化的发展和井下安全生产的加强,以及喷露洒水、煤层注水、综合防尘等措施的实施,原煤水分呈增加的趋势。为此,煤矿除在开采设计上和开采过程中的采煤、掘进、通风和运输等各个环节上制定减少煤的水分的措施外,还应在煤的地面加工中采取措施减少煤的水分。 (1)煤中游离水和化合水 煤中水分按存在形态的不同分为两类,既游离水和化合水。游离水是以物理状态吸附在煤颗粒内部毛细管中和附着在煤颗粒表面的水分;化合水也叫结晶水,是以化合的方式同煤中矿物质结合的水。如硫酸钙(NaSO4.2H2O)和高龄土(AL2O3.2SiO2.2H2O) 中的结晶水。游离水在105~110C的温度下经过1~2小时可蒸发掉,而结晶水通常要在200C以上才能分解析出。 煤的工业分析中只测试游离水,不测结晶水。 (2)煤的外在水分和内在水分 煤的游离水分又分为外在水分和内在水分。 外在水分,是附着在煤颗粒表面的水分。外在水分很容易在常温下的干燥空气中蒸发,蒸发到煤颗粒表面的水蒸气压与空气的湿度平衡时就不再蒸发了。 内在水分,是吸附在煤颗粒内部毛细孔中的水分。内在水分需在100C以上的温度经过一定时间才能蒸发。 最高内在水分,当煤颗粒内部毛细孔内吸附的书分达到饱和状态时,这是煤的内在水分达到最高值,称为最高内在水分。最高内在水分与煤的孔隙度有关,而煤的孔隙度又于煤的煤化程度有关,所以,最高内在水分含量在相当程度上能表征煤的煤化程度,尤其能更好地区分低煤化度煤。如年轻褐煤的最高内在水分多在25%以上,少数的如云南弥勒褐煤最高内在水分达31%。最高内在水分小于2%的烟煤,几乎都是强粘性和高发热量的肥煤和主焦煤。无烟煤的最高内在水分比烟煤有有所下降,因为无烟煤的孔隙度比烟煤增加了。 (3)煤的全水分 全水分,是煤炭按灰分计加中的一个辅助指标。a.煤中全水分的含义。煤中全水分,是指煤中全部的游离水分,即煤中外在水分和内在水分之和。必须指出的是,化验室里测试煤的全水分时所测的煤的外在水分和内在水分,与上面讲的煤中不同结构状态下的外在水分和内在水分是完全不同的。化验室里所测的外在水分是指煤样在空气中并同空气湿度达到平衡时失去的水分(这是吸附在煤毛细孔中的内在水分也会相应失去一部分,其数量随当时空气湿度的降低和温度的升高而增大),这时残留在煤中的水分为内在水分。显然,化验室测试的外在水分和内在水分,除与煤中不同结构状态下的外在水分和内在水分有关外,还与测试是空气的湿度和温度有关。b.煤的全水分测试方法要点见GB212-91。2、煤的灰分 煤的灰分,是指煤完全燃烧后剩下的残渣。因为这个残渣是煤中可燃物完全燃烧,煤中矿物质(除水分外所有的无机质)在煤完全燃烧过程中经过一系列分解、化合反应后的产物,所以确切地说,灰分应称为灰分产率。 (1)煤中矿物质 煤中矿物质分为内在矿物质和外在矿物质。 a.内在矿物质,又分为原生矿物质和次生矿物质。 原生矿物质,是成煤植物本身所含的矿物质,其含量一般不超过1~2%;次生矿物质,是成煤过程中泥炭沼泽液中的矿物质与成煤植物遗体混在一起成煤而留在煤中的。次生矿物质的含量一般也不高,但变化较大。 内在矿物质所形成的灰分叫内在灰分,内在灰分只能用化学的方法才能将其从煤中分离出去。 b.外来矿物质,是在菜煤和运输过程中混入煤中的顶、底板和夹石层的矸石。外在矿物质形成的灰分叫外在灰分,外在灰分可用洗选的方法将其从煤中分离出去。 (2)煤中灰分 煤中灰分来源于矿物质。煤中矿物质燃烧后形成灰分。如粘土、石膏、碳酸盐、黄铁矿等矿物质在煤的燃烧中发生分解和化合,有一部分变成气体逸出,留下的残渣就是灰分。 2SiO2• AL2O3• 2H2O 2SiO2+AL2O3+2H2O↑-→ CaSO4• 2H2O CaSO4+2H20↑-→ CaCO3 CaO+CO2↑”-→ CaO+SO3 CaSO4-→ CaO+SO3 2Fe2O3+8SO2↑-→ 灰分通常比原物质含量要少,因此根据灰分,用适当公式校正后可近似地算出矿物质含量。 (3)煤灰灰分对工业利用的影响 煤中灰分是煤炭计价指标之一。在灰分计加重,灰分是计价的基础指标;在发热量计加重,灰分是计价的辅助指标。 灰分是煤中的有害物质,同样影响煤的使用、运输和储存。 煤用作动力燃料时,灰分增加,煤中可燃物质含量相对减少。矿物质燃烧灰化时要吸收热量,大量排渣要带走热量,因而降低了煤的发热量,影响了锅炉操作(如易结渣、熄火),加剧了设备磨损,增加排渣量。 煤用于炼焦时,灰分增加,焦炭灰分也随之增加,从而降低了高炉的利用系数。 还必须指出的是,煤中灰分增加,增加了无效运输,加剧了我国铁路运输的紧张。 (4)煤的灰分测定见GB212-91。3、煤的挥发分 煤的挥发分,即煤在一定温度下隔绝空气加热,逸出物质(气体或液体)中减掉水分后的含量。剩下的残渣叫做焦渣。因为挥发分不是煤中固有的,而是在特定温度下热解的产物,所以确切的说应称为挥发分产率。 (1)煤的挥发分不仅是炼焦、气化要考虑的一个指标,也是动力用煤的一个重要指标,是动力煤按发热量计价的一个辅助指标。 挥发分是煤分类的重要指标。煤的挥发分反映了煤的变质程度,挥发分由大到小,煤的变质程度由小到大。如泥炭的挥发分高达70%,褐煤一般为40~60%,烟煤一般为10~50%,高变质的无烟煤则小于10%。煤的挥发分和煤岩组成有关,角质类的挥发分最高,镜煤、亮煤次之,丝碳最低。所以世界各国和我国都以煤的挥发分作为煤分类的最重要的指标。 (2)煤的挥发分测试要点见GB212-91。 4、煤的固定碳 煤中去掉水分、灰分、挥发分,剩下的就是固定碳。 煤的固定碳与挥发分一样,也是表征煤的变质程度的一个指标,随变质程度的增高而增高。所以一些国家以固定碳作为煤分类的一个指标。 固定碳是煤的发热量的重要来源,所以有的国家以固定碳作为煤发热量计算的主要参数。固定碳也是合成氨用煤的一个重要指标。 固定碳计算公式: (FC)ad=100-(Mad+Aad+Vad) 当分析煤样中碳酸盐CO2含量为2-12%时: (FC)ad=100-(Mad-Aad+Vad)-CO2,ad(煤) 当分析煤样中碳酸盐CO2含量大于12%时: (FC)ad=100-(Mad+Aad+Vad)-[CO2,ad(煤)-CO2,ad(焦渣)] 式中: (FC)ad——分析煤样的固定碳,%; Mad——分析煤样的水分,%; Aad——分析煤样的灰分,%; Vad——分析煤样的挥发分,%; CO2,ad(煤)——分析煤样中碳酸盐CO2含量,%; CO2,ad(焦渣)——焦渣中CO2占煤中的含量,%;5、煤的硫分 (1)煤中硫存在的形态 煤中硫分,按其存在的形态分为有机硫和无机硫两种。有的煤中还有少量的单质硫。 煤中的有机硫,是以有机物的形态存在与煤中的硫,其结构复杂,至今了解的还不够充分,大体有以下官能团: 硫醇类,R-SH(-SH,为硫基); 噻吩类,如噻吩、苯骈噻吩、硫醌类,如对硫醌、硫醚类,R-S-R' 硫蒽类等 煤中无机硫,是以无机物形态存在于煤中的留。无机硫又分为硫化物硫和硫酸盐硫。硫化物硫绝大部分是黄铁矿硫,少部分为白铁矿硫,两者是同质多晶体。还有少量的ZnS,PbS等。硫酸盐硫主要存在于CaSO4中。 煤中硫分,按其在空气中能否燃烧又分为可燃硫和不可燃硫。有机硫、硫铁矿硫和单质硫都能在空气中燃烧,都是可燃硫。硫酸盐硫不能在空气中燃烧,是不可燃硫。 煤燃烧后留在灰渣中的硫(以硫酸盐硫为主),或焦化后留在焦炭中的硫(以有机硫、硫化钙和硫化亚铁等为主),称为固体硫。煤燃烧逸出的硫,或煤焦化随煤气和焦油析出的硫,称为挥发硫(以硫化氢和硫氧化碳(COS)等为主)。煤的固定硫和挥发硫不是不变的,而是随燃烧或焦化温度、升温速度和矿物质组分的性质和数量等而变化。 煤中各种形态的硫的总和称为煤的全硫(St)。煤的全硫通常包含煤的硫酸盐硫(S
我们在承担农业部的例行监测任务,其中包括测定猪尿中瘦肉精(包括克伦特罗、莱克多巴胺和沙丁胺醇),标准采用农业部1025号公告-11-2008 猪尿中β-受体激动剂多残留检测 液相色谱-串联质谱法。方法里需要使用酶解,比较费时,大家讨论一下酶解和不酶解有多大区别?为什么要酶解?







 我要推广仪器
我要推广仪器
 下载APP
下载APP