怎么分离亚氨基二乙酸和氨基三乙酸,样品中氨基三乙酸含量少
测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
请问做压溶剂峰时刻以压下乙酸乙酯这样同时有两处化学位移的峰吗?谢谢
ICP-AES一般是检测无机元素含量,主要是金属元素。我现有个疑问,亚胺基二乙酸想要检测一下其中的元素含量,并且它可以溶于水,那么水溶解以后是否就可以上机检测了?因曾做过这一类型的有机物,进样到ICP-AES中会熄火,不知道是不是因为有机物的原因,所以不太敢检测亚胺基二乙酸,检测有机物对仪器影响大吗?会不会把仪器烧了?请各位支招。谢谢!
乙酸锌和亚铁氰化钾反应方程式怎么书写,?[color=#333333]亚铁氰化钾可配合乙酸锌作为澄清剂:它是利用乙酸锌与亚铁氰化钾反应生成的氰亚铁酸锌沉淀来挟走或吸附干扰物质。这种澄清剂除蛋白质能力强,但脱色能力差,适用于色泽较浅,蛋白质含量较高的样液的澄清,如乳制品、豆制品等,可以用于可溶性糖类的提取和澄清。[/color][color=#333333][/color][color=#333333]百度上有人这么回答,我想知道具体的反应方程式,或者[color=#333333]氰亚铁酸锌的分子式怎么书写?[/color][/color]
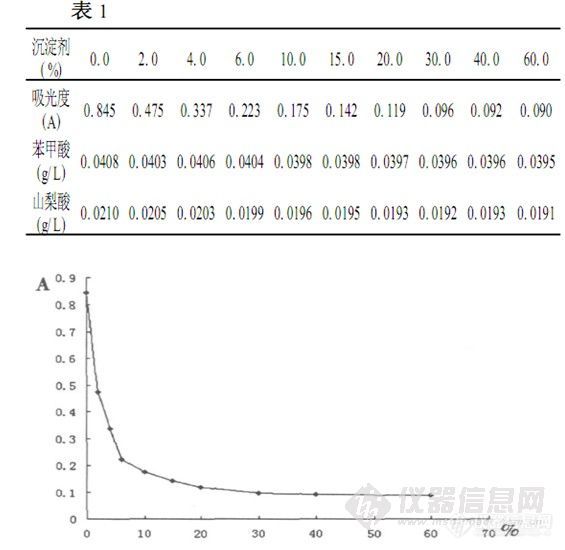
亚铁氰化钾-乙酸锌在 HPLC测定酱油中防腐剂的应用 苯甲酸、 山梨酸作为一种抗微生物剂 ,由于它们对酵母菌和其它细菌的生长抑制很有效 ,且有一定的抗霉菌活性,并且很容易被代谢掉,因此被广泛应用于调味品中 ,由于此类添加剂食用过多会破坏人体肠道微生物平衡 ,对人体健康有一定的伤害。国家食品卫生标准 GB2760 对其用量有明显的限量规定。在按 GB/ T5009、 29 的方法用 HPLC测定其含量时 ,由于酱油含有较多的色素大分子 ,短肽 ,有机酸等大分子颗粒 ,如经一般的稀释、 过滤即上机分析 ,不仅过滤困难 ,而且大分子颗粒极易堵塞色谱柱。造成柱压增大。保留时间变化过大等柱效下降现象。对色谱柱造成难以修复的损伤。使色谱柱使用寿命大大缩短。而食品卫生标准 G B/T5009. 29 - 2003中未对此类样品处理作介绍,因此寻找一种简单而有效的样品前处理方法是非常必要的。在酱油样品前处理过程中,除去大分子颗粒用沉淀法是首选。亚铁氰化钾-乙酸锌作为一种经典的蛋白质及其他大分子极性物质沉淀剂,广泛运用于食品分析中,而且使用方便。在弱酸性条件下使用不受影响。选用此沉淀剂在酱油测定防腐剂过程中进行了一系列试验和应用,经不同浓度沉淀剂沉淀处理后 ,测得的吸光度及防腐剂含量http://ng1.17img.cn/bbsfiles/images/2010/12/201012211713_268817_1638724_3.jpg 沉淀剂用量与吸光度的关系表和图显示沉淀剂的加入量增加而样品处理效果增强,而沉淀剂的加入量对样品中防腐剂含量的测定误差均在允许的误差范围内,但超过20 %的用量时,沉淀处理分离杂质效果明显减小。当亚铁氰化钾2乙酸锌沉淀剂加得较多时,沉淀较多,会给后面过滤沉淀工序操作上带来困难,因此建议亚铁氰化钾2乙酸锌沉淀剂的用量应控制在20 %左右为宜。测定工作中选用量为20 %。
钴(Ⅲ)亚氨基二乙酸的配合物 怎么翻译啊?谢谢了
饮用水一般来源于自来水、桶装水和井水。自来水需经过消毒后才能饮用,其消毒方式一般包括氯消毒(液氯、次氯酸钠消毒等)和二氧化氯消毒。氯消毒因成本低廉的优点,目前是我国大型水厂的主流消毒方式。除卤代烃外,常见的含氯消毒副产物还有亚氯酸盐、氯酸盐、二氯乙酸和三氯乙酸等。这四种消毒副产物目前成为生活饮用水的常规检测项目,因此如何分离这四种消毒副产物成为目前一大热点。本文探索并开发[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]分离亚氯酸盐、氯酸盐、二氯乙酸和三氯乙酸的方法。首先是色谱柱和定量环的选择。由于一般饮用水中亚氯酸盐、氯酸盐、二氯乙酸和三氯乙酸在水中的检测值较低,因此需要采用大定量环测定,选择定量环体积为是500μL。实验所用 IonPac AS19分离柱亲水性好,柱容量高,能够满足生活饮用水中常见离子、卤乙酸及卤氧乙酸的同时测定。选择KOH作为淋洗液,利用淋洗液在线发生技术实现梯度洗脱,经过 AERS4 mm自动再生微膜抑制器抑制后产物为水,背景电导低,水负峰不明显,能够实现大体积进样,显著提高方法的灵敏度。淋洗液梯度的选择。选择以初始浓度分别为8、12、15 mmol/L来进行实验,结果表明,当初始浓度为8 mmol/L时,分离效果较好,使用初始浓度为12 mmol/L时,三氯乙酸受到硫酸盐的前延展性峰的干扰,分离效果不好;当浓度为15 mmol/L时,出峰速度较快,四种消毒副产物分离效果不好。由于三氯乙酸极性较大,需要采用梯度淋洗方法将进行洗脱。当选择(20-32)min匀速升至25 mmol/L,保持2 min,可以将保留时间较长的三氯乙酸尽快洗脱出来,且分离效果较好,减少检测所用时间,增加方法的实用性。梯度洗脱程序如表[align=center]表 四种消毒副产物的梯度洗脱程序[/align][table][tr][td]时间(min)[/td][td]梯度浓度C[sub]NaOH[/sub](mmol/L)[/td][/tr][tr][td]0-20[/td][td]8[/td][/tr][tr][td]20-32[/td][td]8-25[/td][/tr][tr][td]32-34[/td][td]25[/td][/tr][tr][td]34[/td][td]8[/td][/tr][/table]实际样品的测定。先对预先活化Ag柱、Ba柱和H柱,分别用注射器以2 mL/min 的流速将10mL超纯水过柱,静置10 min使其充分平衡。然后直接取适量水样,以2 mL / min的速度依次通过串联的Ag 柱、Ba 柱、H柱和0. 22 μm针式滤器,弃去前面 6 mL后开始收集滤液,滤液直接进样测试。可以明显去除氯离子和硫酸盐的含量,减少干扰峰的影响。实验中注意事项和建议先使用标准溶液分离这四种消毒副产物,再对三氯乙酸加标水样进行测定分离,确保三氯乙酸和硫酸盐可以有很好的分离度。二氧化碳装置使用。如果装有二氧化碳装置会大大降低硫酸根前延展性峰的干扰。使分离效果更好。
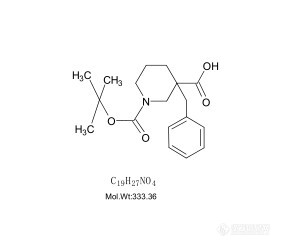
色谱世界的各位大侠们,1-boc-3-苄基哌啶甲酸文献报道说用4.6mm*150mm C18的二氧化硅柱子,用乙腈、水、三氟乙酸作为流动相,在214和254处有吸收峰。我们用紫外全波长扫描后,发现只有在204处有紫外吸收峰,可是我们用乙腈和水作流动相,这个东西在214处不出峰。这个东西该怎么检测纯度呢。这个化合物是通过1-boc-3-哌啶甲酸 和溴苄合成的, 还有1-boc-3-哌啶甲酸 的熔点是159-162℃。 我们这个东西的熔点是109-116℃。 所以这两个东西很定有是不一样的。 实在不行只有打核磁了。我们这个原料的紫外吸收也在204这个位置。但我们用254的紫外薄层检测,发现原料不显色,1-boc-3-苄基哌啶甲酸 轻微显色。[img=,281,247]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231115067664_5937_1815404_3.jpg!w281x247.jpg[/img]
想问问大神,乙酸锌亚铁氰化钾除蛋白的步骤是怎样,加入各1mL的乙酸锌和亚铁氰化钾,下一步有的说弃去初滤液是什么意思,滤液备用,滤液是哪部分,还有说弃去有机层,水层和有机层哪个在上面?
IC-测定饮用水中亚氯酸盐、氯酸盐、溴酸盐、二氯乙酸及三氯乙酸
我要做氨基三乙酸和亚氨基二乙酸的分离,但是在C18柱上试了好几种方法都没有保留,请专家帮忙
大家好,请问近红外是否可以测定蛋白质亚基含量,就是想建立SDS-PAGE测定的蛋白质亚基的快速检测方法,请各位指点,谢谢!
有哪位朋友做过哌啶基哌啶的分析,能否跟我联系下,有一些问题想请教谢谢
问题: 我想请教下,亚铁氰化钾和乙酸锌做沉淀剂的时候,是要哪个先加吗回复: 亚铁氰化钾先
因工作需要,急需亚氨基二乙睛、亚氨基二乙酸化学法分析方法。我手里已有高效液相色谱分析方法,但单位没有液相色谱设备,只能干着急啊。
请教 氯乙酸 苯酚 苯氧乙酸用什么做展开剂?即氯乙酸和苯酚反应生成苯氧乙酸。
乙酸对叔丁基环己酯 和乙酸邻叔丁基环己酯如何区分?谢谢
2010年版药典氟哌啶醇片红外鉴别方法是:取本品粉末适量(相当氟哌啶醇约50mg),用20ml三氯甲烷分次研磨使溶解,过滤,取滤液水浴蒸干,残渣减压干燥后,压片,与标准图谱比较. 因为氟哌啶醇易溶于三氯甲烷,我在做的过程中就没有剥片;另氟哌啶醇对光 热具不稳定性,滤液采用60度水浴蒸至近干,采用药典原料项下的方法干燥:即60度减压干燥,约5小时,但取出压片时发现它的性状为半固体,压出的片透明度不好,还粘模具,没法做! 会不会是辅料有影响呢?想请教一下,如何改进我的试验,压制一个好的片子?
亚铁氰化钾和乙酸锌去蛋白,去蛋白后过滤进质谱可以吗?
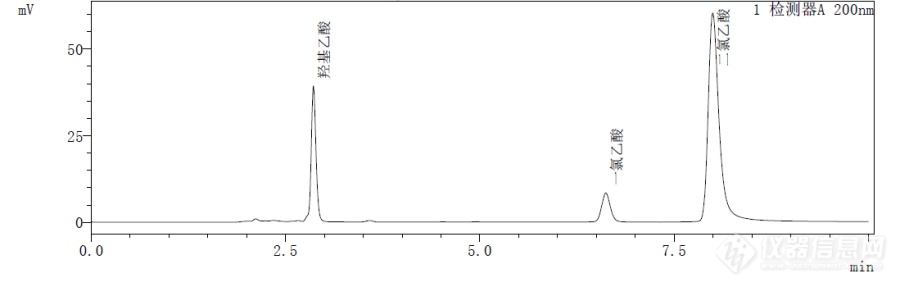
[align=center][b]椰油酰胺丙基甜菜碱中一氯乙酸、二氯乙酸和羟基乙酸的测定[/b][/align] 椰油酰胺丙基甜菜碱(CAB)是一种两性表面活性剂,因其对眼睛和皮肤刺激性低,对头发和皮肤有护理效果并产生大量稳定泡沫,在肥皂和硬水中有出色的起泡性和洗涤性,故广泛用于香波和泡沫浴液等洗涤用品中。 在工业生产中,常使用一氯乙酸(MCA)作为原料生产CAB。而工业MCA中含有少量的二氯乙酸(DCA),DCA是生物学证实具有潜在致癌风险的物质,同时在生产过程中残留的MCA对皮肤、黏膜有很强的腐蚀性,通常采用水解法将MCA转化为刺激性更小的羟基乙酸(GCA)。椰油酰胺丙基甜菜碱产品的指标含量分析中,一般要求一氯乙酸<20ppm,二氯乙酸<300ppm,羟基乙酸<0.5%。[b]色谱条件:[/b]色谱柱:[b]Kromasil C8(4.6*250mm,5μm)[/b]柱 温:24℃检测器:紫外检测器波 长:200nm流动相:乙腈:水=10:90(每1000mL中加入2.0mL磷酸)流 速:1ml/min进样体积:20μL采集时间:10min[img=,690,219]https://ng1.17img.cn/bbsfiles/images/2018/10/201810291003374445_9066_2428063_3.png!w690x219.jpg[/img] 图1 :一氯乙酸、二氯乙酸和羟基乙酸混标色谱图[img=,690,328]https://ng1.17img.cn/bbsfiles/images/2018/10/201810291003547039_780_2428063_3.png!w690x328.jpg[/img] 图2 :椰油酰胺丙基甜菜碱样品色谱图[b]总结[/b]参考国标GB/T 28193-2011表面活性剂中氯乙酸(盐)残留量的测定方法,建立高效液相色谱法,一次性测定样品中一氯乙酸、二氯乙酸和羟基乙酸的含量。其优点是以高比例水相作为流动相,样品不需要进行萃取、酯化等前处理,操作方便,快速高效。使用Kromasil C8色谱柱分离样品中一氯乙酸与其余组分,效率高,分离度好,结果可靠,可为椰油酰胺丙基甜菜碱生产厂家提高产品质量提供参考。[b]注:由深圳爱湾医学检验实验室验证 [/b]
请问测定苯甲酸山梨酸中处理样品时,亚铁氰化钾和乙酸锌顺序为什么不能颠倒
以前很少做气相,现在要做溶剂残留,主要是测乙酸乙酯,仪器是岛津2014,顶空进样。有以下几个问题,求助各位,先谢谢啦1、溶剂应该选什么,我们目前是用二甲亚砜做溶剂,是否可以,二甲亚砜的沸点比较高,应该比乙酸乙酯出峰要晚吧2、大家有没有用内标,是不是自动进样就可以不用内标呢?3、用什么色谱柱合适,我用的是HP-5的柱子4、气相做样品时,每做完一针是否需要在初始温度平衡一段时间
氯乙酸和苯酚反应生成苯氧乙酸选什么展开剂?
最近在做一个原料药的残留溶剂测定,其中有乙酸乙酯,甲醇,DMF,冰乙酸等残留溶剂需要测定。经过摸索后选用HP-innowax柱测定这四种残留溶剂,用水做溶剂配制对照品溶液和供试品溶液,直接进样测定。可是乙酸的峰型时好时坏,不知道是什么原因?大家都用什么方法与柱子测定乙酸,已经试过DB-624与HP-5柱,乙酸在这两根柱子上的峰型都不好,请大家帮帮忙吧,试了好久了,都没解决
哪位好人有三氟乙酸、N-羟基琥珀酰亚胺、二环己基碳二亚胺的质量标准啊?请大家帮助。
昨天做实验发现 往浓硫酸加入乙酸乙酯时 没有分层而是乙酸乙酯全部溶到浓硫酸里面。这时什么原因呀?浓硫酸能氧化乙酸乙酯吗?请高手指教?谢谢!
各位大神,求指点!做脱氢乙酸有情况。按GB/T23377做的,标样很好,线性6个9,但是样品加标回收低,只有50-60%。流动相PH大概是6-7,0.02mol乙酸铵:甲醇=90:10。做了两组样品,一组是饮料加标,样品离心后没有过萃取小柱,样品PH调成4-5一组是面包加标,样品用亚铁氰化钾和乙酸锌除蛋白,样品离心后用正己烷萃取除脂肪,样品PH调成4-5加标的结果不对啊,饮料加标25ug定容到25ml,看不出什么峰形,几乎检不出来,加标600ug定容到25ml,回收率只有63.75%面包加标25ug定容到25ml,几乎检不出来,加标600ug定容到25ml,回收率只有55%目标峰有拖尾,但不至于这么影响回收率吧?!
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先[em0813] [em0813] [em0813]补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先 补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论跟上次的问题一样,上次结贴太匆忙了,再次征集。感谢上次renture/jiangyunjun3/melu的帮助,找到一个分光法测痕量氮川的方法,测量范围是0.01~0.12ug/mL,但我们的样品是含量大概有0~10%左右的氮川,是否能够通过稀释来使用上面的分光方法还不知道。现在想征集一个能够分析常量氮川的方法,希望大家踊跃发言。







 我要推广仪器
我要推广仪器
 下载APP
下载APP