聚山梨酯80残留溶剂检测中的(三乙胺与二甲基甲酰胺),三乙胺的出峰面积越来越大。不知道大家有没有碰到过这种情况,出峰面积一针比一针大,求指点!
请问一下哦,我在做完砷化氢测定实验时, 二乙胺基二硫代甲酸银-乙醇氯仿溶液吸收完后,用乙醇清洗吸收管,乙醇在里面放置一夜后,吸收管理出现类似白色的亮晶晶的小片状固体,很想知道这是什么东东啊,请各位指点一下,谢(⊙o⊙)哦
谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法
求助无水硫酸镁,氢化钠,氯甲酸乙酯,四氢呋喃,乙腈(hplc),N-甲基吗啉,二异丙基乙胺,对甲苯磺酸的质量标准。大家有哪个给我哪个就可以。这些资料库里都没有。在外面也实在找不到了。大家帮忙。
测二甲基,乙醇,三乙胺用什么柱子比较好?
请问:二甲基疏基乙胺国内有否厂家生产此产品?能否提供相关的分析此产品的信息?
顶空[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]做残留溶剂三乙胺的回收率为什么会降低?具体如下:第一针进溶剂5毫升二甲基亚砜,第二针进标液(500mg三乙胺用二甲基亚砜定容100毫升,取5毫升用二甲基亚砜定容到59毫升),第三针称0.5g样品加二甲基亚砜5毫升,第四针称0.5g样品加5毫升第二针的标液。
各位大师,快帮帮忙啊。做二硫化碳时,二乙胺需要提纯,查了些资料说是要先加入甲苯磺酰氯,乙胺与二乙胺沉淀下来,过滤掉三乙胺;再在沉淀中加入氢氧化钠,乙胺反应掉,二乙胺不反应。但问题是我需要的是二乙胺溶液啊,怎么将二乙胺从沉淀中分离出来呢
各位大师,快帮帮忙啊。做二硫化碳时,二乙胺需要提纯,查了些资料说是要先加入甲苯磺酰氯,乙胺与二乙胺沉淀下来,过滤掉三乙胺;再在沉淀中加入氢氧化钠,乙胺反应掉,二乙胺不反应。但问题是我需要的是二乙胺溶液啊,怎么将二乙胺从沉淀中分离出来呢
做溶剂残留,用的月旭的wm624色谱柱,混合对照其他峰都有,就三乙胺没有峰,溶剂是nn二甲基甲酰胺,顶空进样
要测一个原料药中三乙胺的残留。仪器是安捷伦的6890N,7694E,条件如下:色谱柱HP-1,柱温100,进样口250,分流比1:1,柱压7.7psiFID250顶空平衡30min,温度70/100/110三乙胺浓度16ug/ml(用水溶),取2ml至10ml顶空瓶我的问题是我进了三乙胺后跟了一针空白,三乙胺的位置有出峰,再跟一针空白就没有了,我试着做进样精密度,连续进了三针峰面积从29涨到了40,RSD非常差。而且换了DB-624的柱子,这个问题还是存在,哪位大侠指点一下!
空气二硫化碳的测定中二乙胺需要蒸馏,有没有大佬可以告诉具体的蒸馏方法?最好详细一点,谢谢
有什么比较好的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]方法将六甲基二硅氧烷(硅醚)和三乙胺分开
利用顶空-[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测试三乙胺含量,感觉前后两针互相干扰,进完一针样品后,再进空白,会有三乙胺的峰,然后再进一次空白三乙胺的峰面积会减小,应该是三乙胺残留了,请问有什么办法让三乙胺不残留。
二硫化碳的二乙胺分光光度法,中的吸收液配置出来怎么是蓝色 ,加入氨水后就变蓝了 ,,,,,,你们都是什么色吗
我做了三次曲线,都是0.99不达0.999,到底是什么原因呢?二乙胺需要蒸馏吗?求老师解答,谢谢。
我在做二硫化碳的标准曲线,执行标准为GBZ/T160.33-2004中的二硫化碳的二乙胺分光光度法,做了几次,显色为黄棕色,但没有梯度,而且空白管也显色,不符合标准,(空白应为无色),各试剂重新配过了,找不出问题所在,哪位高手指点,谢谢。
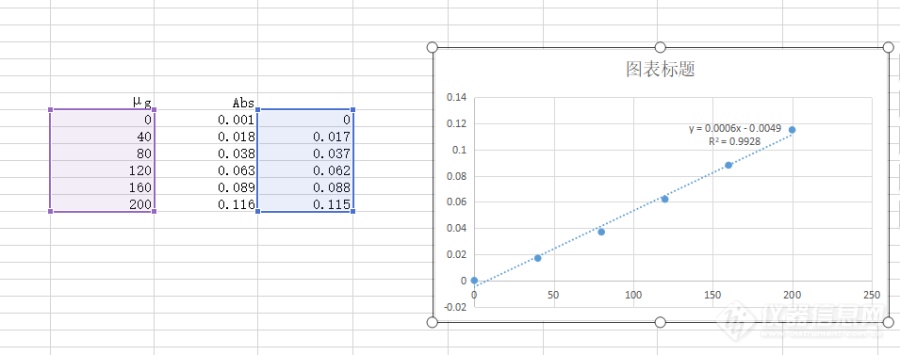
有没有二硫化碳的标准曲线参考一下?我做的曲线吸光度很低,线性也很差,二乙胺和三乙醇胺都是分析纯,二乙胺没有提纯,用的科密欧的无苯级二硫化碳配的标液,配制过程是称取0.0400g二硫化碳用无水乙醇定容到50ml容量瓶配成储备液,再取5ml储备液用无水乙醇定容到50ml容量瓶,算出来标准使用液的浓度是80μg/ml,我试过配成10μg/ml的使用液做曲线但是吸光值很低没有颜色梯度,这一次配成80的浓度勉强能看出点颜色梯度但是线性也不好。。[img=,690,272]https://ng1.17img.cn/bbsfiles/images/2022/01/202201181847008977_5382_3515878_3.png!w690x272.jpg[/img]
空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量二硫化碳二乙胺标线截距一直是负的 还只能做到两个九 请问各位老师有什么解决办法吗
最近测一个水溶性样品的三乙胺和其他有机溶剂的残留,被三乙胺搞得有点小郁闷。之前用直接进样的方法,用水做溶剂,中等极性的毛细管柱,三乙胺出了一个馒头峰,峰很宽,灵敏度很低,不成线性。听说是三乙胺和水氢键结合了。后来换成用DMSO或DMF溶,灵敏度提高很多,但样品不溶于这俩溶剂中。于是打算用顶空进样,还是用水溶,试问这样能否改善三乙胺的灵敏度问题,比直接进样好些?能摆脱水对三乙胺的干扰?
我做样品的三乙胺残留验证时,总是不成线性,有谁做过这个溶剂的,告知下方法,谢谢。
三乙胺残留溶剂做方法学验证的线性时,浓度和峰面积不成正比,液体进样,DMF做溶剂

如题,原料药合成中,溶剂用到三乙胺,那么三乙胺作为溶剂残留,质量标准应该定多少合适?查了ICH,把三乙胺归到了3类溶剂里,标准推算如下图:[img=,690,504]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091239303097_6773_2700892_3.png!w690x504.jpg[/img]6250ppm是根据毒理学数据计算的结果。但查了相关欧盟方面的规定,三乙胺定了320ppm,结合以上信息,那三乙胺作为溶残到底应该定多少比较合理?
我在做某个产品的溶剂残留,里面有甲醇,乙醚,三乙胺,DMF,乙腈和二氯甲烷。因为DMF用顶空做的话,响应太低,用直接进样响应会高些。目前我使用DB-624, DB-5的柱子三乙胺都做不好,峰型太拖,用非极性和弱极性的柱子做的话,甲醇,乙醚,乙腈等分离度又太差。有什么办法使这些溶剂残留能在一根柱子里出峰呢?向大家请教。
最近在做溶剂残留检测,一共六种残留溶剂做了一个混标的方法,其中二乙胺跟DMF结果不理想,首先配的混标溶液连续进样5次,二乙胺跟DMF回收率都很好,然后做了样品加标的准确度试验,二乙胺回收率低于70,DMF重复性不好,而且高于120,使用的是7890A,顶空进样,柱子是DB624,60米的柱子,但是二乙胺跟正己烷前后出峰,二乙胺有点拖尾,请教大家怎么能改善??问题在哪里?
最近在做三乙胺和乙腈的残留,用乙醇做内标,水做溶剂,DB-624(30m 0.32mm 1.8um),不分流,进样量0.4ul,进样口200度,检测器250度,恒温40度。在此方法下,三乙胺可以完美出峰,乙腈和乙醇的峰均有开叉。有没有大佬懂,可以改变哪一个条件,能做出来(除了顶空,没有安装顶空)。拜谢![img]https://ng1.17img.cn/bbsfiles/images/2022/04/202204121619588907_9684_5121239_3.png[/img]
我在做二硫化碳的检测标准曲线,执行标准为GBZ/T160.33-2004中的二硫化碳的二乙胺分光光度法,做了几次,颜色反应是黄棕色,但是吸光度值是一样的,空白管也显色,不符合标准,(空白应为无色),请问可能是哪里出现问题呢?谢谢。
三乙胺怎么除水呢,我要求不高,查资料说不能用硫酸镁,可以用氯化钙吗
求三乙胺溶剂残留气相检测方法
哪位大佬知道苯酚,乙胺这两个在残留溶剂测定时候的限度为多少







 我要推广仪器
我要推广仪器
 下载APP
下载APP