CAR-T研究前沿应用|珀金埃尔默与您相约第十九届中国生物制品年会(CBioPC2019)
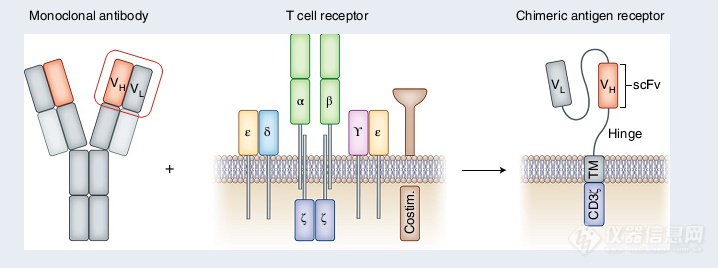
背景介绍: CAR-T疗法风潮基于Chimeric antigen receptor T cells (CAR-T)技术的细胞治疗可谓是当下最火热的肿瘤治疗方法之一。与早期的基于T细胞的Adoptive cell transfer(ACT)不同,CAR-T技术融入B细胞,也就是抗体的识别能力。因此CAR-T能靶向MHC异常表达或不表达的肿瘤细胞,并能作用于多数传统ACT无法靶向的细胞膜表面靶点[1]。CAR-T原理图,来源于参考资料[1]随着在B细胞血液肿瘤领域的巨大成功,CAR-T疗法的临床前研究和临床试验发展迅速。目前,就临床数目而言,CAR-T疗法在肿瘤免疫领域仅次于基于免疫检查点抑制剂的疗法。中国和美国则是推动CAR-T疗法临床转化的主力军[2]。相较于美国,接近八成的国内研究已处于临床阶段。同时,对比2018年,我们可以看到国内制药企业对于CAR-T疗法的布局和推动[2]。CAR-T治疗临床研究数目,来源于参考资料[2]当下,如何将CAR-T疗法的成功复制至非B细胞血液肿瘤和实体瘤是该领域研究的关键点和重点。在实体瘤研究方向,体内研究和临床试验证明Therapeutic window的存在,并发现CAR-T细胞能清除靶点阴性的肿瘤细胞,为攻克实体瘤提供一丝曙光[2]。除了肿瘤领域,CAR-T研究已衍生至心脏损伤[3],HIV[4]和自身免疫[5]等多个疾病领域,成为了非常有潜力的新兴治疗技术。在此,结合一线的研究进展,我们向大家介绍珀金埃尔默生命科学产品线针对CAR-T研究的应用切入点,助力下一代CAR-T疗法研发。体外研究应用:针对CAR-T研究,珀金埃尔默主要围绕体外和体内两个方向提供完善的解决方案。其中,针对体外研究,我们关注体外CAR-T细胞的活化、活力检测和分析。与Cytotoxic T细胞活化类似,CAR-T细胞的活化通常伴随着细胞因子的分泌、CAR-T细胞的增殖和最后特异性靶向肿瘤细胞的杀伤 [6]。针对这三个过程,我们均提供对应的基于放射/非放射平台的金标准解决方案。在此,我们关注CAR-T细胞核心功能指标:细胞杀伤的检测。1. 灵活应用细胞杀伤解决方案与传统的基于化合物和抗体的直接细胞杀伤不同,基于CAR-T细胞的肿瘤杀伤是细胞-细胞相互作用的结果。因此,在分析杀伤中,检测方法必须能够特异区别靶细胞(肿瘤细胞)和效应细胞(CAR-T细胞)。常见的细胞活力检测法,如MTT、CCK-8和ATP法等,则不适于直接用于CAR-T介导的特异细胞杀伤检测。针对区分的需求,我们提供两大金标准检测法:基于放射的51Cr释放法和基于非放射的DELFIA® EuTDA细胞毒法[7]。两种方法都基于利用探针高效、特异的标记靶细胞。经效应细胞杀伤后,靶细胞出现裂解,并释放探针到上清中用于定量杀伤效果。51Cr释放法检测原理图,图片来源于参考资料[8]除了支持特异检测细胞杀伤外,51Cr释放法和 DELFIA® EuTDA细胞毒法的另外一大优势就是灵活性,支持多种细胞系和原代细胞作为靶细胞或效应细胞。例如,在近期的靶向CD19的CAR-T临床研究中,科研人员通过标记肿瘤细胞,利用51Cr释放法证明低亲和力的CAR-T 具有更强的细胞杀伤能力。而在后续的CAR-T无效的病人样本研究中,基于同样的检测方法,科研人员通过标记CAR-T细胞作为靶细胞,检测病人针对CAR-T细胞是否具有免疫反应(Anti-CAR反应)[9]。 体外杀伤检测结果。CAT为低亲和力CAR-T,FMC83为常用CAR-T 基于病人临床样本的Anti-CAR检测结果。图片来源于参考资料[9]通过改变靶细胞,我们提供的方法学还支持CAR-T疗法的安全性分析,例如检测On target, off tumor脱靶效应带来的细胞毒性[10]。此外,同种异体反应也逐渐成为CAR-T疗法安全性评价的一个重要考虑因素。针对此,我们提供DELFIA® BrdU 细胞增殖方法,通过混合淋巴细胞反应衡量CAR-T细胞的同种异体反应[11]。利用51Cr释放法检测脱靶效应的细胞毒性,图片来源于参考资料[10];基于DELFIA® BrdU 细胞增殖方法检测CAR-T细胞的同种异体反应,图片来源于参考资料[11]2. 深入成像技术应用相较于分子检测,成像技术能提供二维到三维的静止或动态空间信息。因此基于成像的高通量筛选更具有选择性,适合差异化筛选。例如,在2017年的临床研究中,科研人员利用Opera Phenix高内涵平台,结合肿瘤细胞/正常细胞混合样本进行差异化筛选。利用成像技术的优势,研究能精确发现只作用于肿瘤细胞、不影响正常细胞活力的药物,提升了临床用药的安全性[12]。除了提升选择性外,成像筛选还能支持几小时到几天的长程筛选,因此可用于检测免疫细胞的多轮杀伤能力[13]。基于Opera Phenix 的高通量差异筛选流程图,图片来源于参考资料[12]毫无疑问,凭借其生理相关性和支持高通量筛选的优势,3D类器官和微组织球研究是当下多个科研和制药领域的热门主题和方向,也是我们成像应用的重要关注点。针对多种肿瘤,基于小分子药物的研究已证实3D肿瘤组织样本能有效预测药物敏感度[14]。在CAR-T和细胞治疗研究方向,3D水平研究也逐渐崭露头角。在近期的研究中,Opera Phenix高内涵平台被用于衡量、对比靶向Mesothelin的CAR-T细胞的杀伤能力 [15] 。除了3D水平研究外,活细胞动态监测也会成为我们针对CAR-T研究的主要开拓点。与干细胞类似,CAR-T细胞和免疫细胞在活化和行使功能过程中是代谢水平高度动态的,并持续存在和癌细胞及其他免疫细胞的相互作用 [16]。因此,基于成像的动态分析非常适合在体外深入解析CAR-T细胞的功能潜能。利用Opera Phenix 检测CAR-T细胞针对3D肿瘤样本的杀伤能力,图片来源于参考资料[15]体内研究应用:针对体内研究,珀金埃尔默主要提供完善的活体成像解决方案,包括性能卓越的IVIS系列和对应的细胞株和荧光染料。作为主流的活体成像平台,IVIS系列被广泛用于临床前CAR-T研究和突破实体瘤治疗的多种瓶颈[1]。在此,我们主要介绍助力下一代CAR-T研发的两大利器:肿瘤成像和免疫细胞成像。1. 肿瘤活体成像:协助建立复杂的肿瘤模型围绕CAR-T临床前研发,IVIS系列能协助建立多种常见的实体瘤和血液瘤模型,灵敏追踪肿瘤的进展和动态评估CAR-T细胞的抗癌作用。在此基础上,IVIS系列还可以和多种新兴的技术联用,协助建立复杂的肿瘤表型。例如,在靶向多发性骨髓瘤的CAR-T临床治疗中,一个常见的挑战就是BCMA阴性肿瘤细胞的存在和出现。对此,研究结合Crispr技术,建立融合萤火虫报告基因的BCMA阴性肿瘤细胞,并将其混合BCMA阳性肿瘤细胞建立肿瘤模型。活体成像数据可以看出,BCMA CAR-T细胞无法作用于BCMA阴性肿瘤细胞,而靶向GPRC5D的CAR-T细胞能有效根除BCMA阴性肿瘤细胞[17]。结合Crispr技术的肿瘤模型建立和活体成像检测,图片来源于参考资料[17]同样基于Crispr技术,近期的研究利用活体成像平台开展和验证靶向CD8 T细胞的活体Crispr筛选,为靶向胶质母细胞瘤的免疫治疗提供了新的靶点[18]。通过改变接种的流程和治疗策略,IVIS系列还可以协助建立肿瘤复发、晚期肿瘤和病人来源移植肿瘤模型。基于Crispr技术的体内筛选验证流程,图片来源于参考资料[18]2. 免疫细胞活体成像:直观展示体内免疫细胞-肿瘤细胞相互作用 除了肿瘤细胞成像,我们还能通过标记CAR-T细胞或其他免疫细胞进行活体成像[17]。该应用对于CAR-T和其他免疫疗法的研究尤为重要。因为通过活体成像,我们能直观的监测、分析免疫细胞的体内动态分布和迁移等关键的信息。鉴于CAR-T细胞体内的扩增、迁移和肿瘤部位的杀伤功能是当下CAR-T临床应用的主要限制因素[1]。这些信息的获得对于新一代CAR-T研发至关重要。在近期的一份研究中,科研人员展示了CAR-T细胞迁移能力对于杀伤效果的重要性[19]。利用肿瘤细胞,尤其是在辐射处理后,会过表达IL-18的特点,研究人员改造CAR-T细胞,使其过表达IL-18受体 CXCR-1或CXCR-2来提升体内的迁移能力。结合带有报告基因的CAR-T细胞和过表达远红外蛋白iRFP720的肿瘤细胞,研究就能直观追踪和对比免疫细胞的分布变化和肿瘤的进展情况,做到体内水平的细胞细胞相互作用研究。相较于对照细胞,改造的CAR-T细胞具有更强的迁移能力和杀伤肿瘤能力。同时,在晚期肿瘤模型上,研究证实未改造CAR-T细胞主要分布于外周,不能有效靶向肿瘤部位[19]。CAR-T体内模型的CAR-T细胞成像(左,化学发光)和肿瘤细胞成像(中,荧光);右图为对应统计数据,图片基于参考资料[19]进一步结合免疫荧光技术,研究证实了由IVIS采集的化学发光信号能有效反映治疗早期的CAR-T细胞浸润情况,是肿瘤微环境免疫细胞活体追踪的有力手段。同时,治疗早期的化学发光信号也能有效预测小鼠的生存时间,为CAR-T疗效预测提供了新的切入点。最后,同时结合肿瘤和CAR-T细胞活体成像,我们还能做到靶向CAR-T细胞功能水平的持续监测。在首次肿瘤消退后,我们再次植入肿瘤细胞,建立Re-challenge模型,通过活体成像直观评价CAR-T细胞的长期分布和功能[19]。利用免疫荧光和活体成像技术追踪治疗早期CAR-T细胞的侵润情况;CD45指示CAR-T细胞,图片来源于参考资料[19]参考资料:1. Robbie G. Majzner and Crystal L. Mackall?.Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019 Sep 25(9):1341-1355.2. Jia Xin Yu, et al. The global pipeline of cell therapies for cancer Nat Rev Drug Discov. 2019 Oct 18(11):821-822.3. Haig Aghajanian, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019 Sep 573(7774):430-433.4. Herzig E, et al. Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell. 2019 Oct 31 179(4):880-894.e10.5. Taking CAR-TCells Beyond Cancer: A New Therapy for Autoimmune Disease https://www.labiotech.eu/interviews/car-t-cells-txcell-treg-cells/6. Restifo NP , et al. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012 Mar 22 12(4):269-81. 7. 细胞治疗干货 | 免疫细胞杀伤经典案例https://mp.weixin.qq.com/s/47krDPy-vsxS5AP91T1GDw8. van der Haar àvila I , et al. Evaluating Antibody-Dependent Cell-Mediated Cytotoxicity by Chromium Release Assay. Methods Mol Biol. 2019 1913:167-179.9. GhorashianS , et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treatedwith a low-affinity CD19 CAR. Nat Med. 2019 Sep 25(9):1408-141410. Fisher J , et al. Engineering γδT cells limits tonic signaling associated with chimeric antigen receptors. Sci Signal. 2019 Sep 10 12(598).11. Chan WK, et al. Chimeric antigen receptor-redirected CD45RA-negative T cells have potent antileukemia and pathogen memory response without graft-versus-host activity. Leukemia. 2015 Feb 29(2):387-95.12. Snijder B, et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017 Dec 4(12):e595-e606.13. Hernandez-Hoyos G, et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of MetastaticCastration-Resistant Prostate Cancer. Mol Cancer Ther. 2016 Sep 15(9):2155-65.14. Lee JK, et al. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy;Nat Genet. 2018 Oct 50(10):1399-1411.15. Zhang Z, et al.Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 2019 Jun 17 10(7):476.16. Sukumar M, et al.Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol. 2017 Jun 46:14-22.17. Smith EL, et al.GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019 Mar 27 11(485).18. Ye L, et al.Invivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol. 2019 Nov 37(11):1302-1313.19. Jin L, et al.CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019 Sep 5 10(1):4016.更多最新资讯及用户交流敬请关注CBioPC2019盛会第十九届中国生物制品年会(CBioPC2019)将在北京国际会议中心举行。本届年会适逢建国七十周年大庆和中国生物百年华诞,特以“展示新中国生物制品七十年成就、回顾中国生物制品百年历史传承”为主题,开展隆重、热烈和丰富多彩的学术交流,产业交流和行业交流。珀金埃尔默公司作为生物制药行业的仪器&试剂供应商,一直致力于为生物药物研发的科学家们提供全方位的应用方案、优质的服务和开放的交流平台,会议期间珀金埃尔默将带来精彩报告,报告内容皆干货,展台活动亦精彩,期待在D07展位与您见面。报告题目:从基因到表型组学的药物研发一体化智能平台报告人:珀金埃尔默市场开发主管 徐雍羽博士报告时间:11月30日9:20-9:45报告地点:细胞治疗与基因治疗分会场长按识别二维码进行注册,并参与抽奖





























 我要推广仪器
我要推广仪器
 下载APP
下载APP