求助,哪位大神做过2020版中国药典的氢化蓖麻油脂肪酸组成呢?最近在做确认,配制的对照品溶液连续进样6针,12-羟基硬脂酸甲酯峰峰面积峰形都相近,RSD很小,但是随行的对照品溶液在进了6针样品后峰形变矮且拖尾,峰面积也差了好几倍,有做过的这样的吗?
求助,有哪位大神做过2020版中国药典的氢化蓖麻油脂肪酸组成呢?最近在做这个,配制的对照品溶液连续进样6针,12-羟基硬脂酸甲酯峰峰形和峰面积都很相近,但是在进样6针样品后的随行对照品溶液,12-羟基硬脂酸甲酯峰的峰形变矮且拖尾,峰面积也变小了一半多,更换了衬管后刚开始峰面积正常,进了样品后就不行了,有什么解决方法吗
药典蓖麻油含量测定 供试品制备里,如图绿色部分应该是在氢氧化钾的催化下和甲醇进行酯交换反应,但如图红色部分加三氟化硼乙醚-甲醇(1:3)是什么目的?求指教
网上查,蓖麻油的分子量很大 不知道用气质能否检测是否出峰?
如题:哪位版友有蓖麻油的具体检测方法及标准?盼应助!
求蓖麻油含量测定,供试品含量公式,还有大神们怎么制备的供试品的,为啥我做出来的含量只有20%左右
大家好!小弟是做化妆品原料检测的,在测量氢化蓖麻油(12-羟基-十八烷酸甘油酯)的碘值时,溶剂采用氯仿,测出的结果比供应商COA文件给出的大,但供应商说是方法不同,他们的溶剂用的CCl4,并且加了乙酸汞,测出的结果正好在范围之内,我想问用这两种溶剂测出的碘值有啥不同?还有乙酸汞的加入是不是只起到缩短检测时间的作用,对最终的结果并无影响??
各位兄弟姐妹们: 有谁了解聚蓖麻油酸、四聚蓖麻油酸,他们的是怎么产生的,有什么区别等等,越详细越好!先谢谢啦!
如题:蓖麻油酸中的苯酚怎么检测含量?如果直接进入气相色谱,植物油酸的汽化点太高,不宜直接汽化。
如题:蓖麻油酸中的苯酚怎么检测含量?如果直接进入气相色谱,植物油酸的汽化点太高,不宜直接汽化。
如题:蓖麻油酸中的苯酚怎么检测含量?如果直接进入气相色谱,植物油酸的汽化点太高,不宜直接汽化。
最近在做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]氢化蓖麻油脂肪酸组成检验,有做过的吗?
各位,请问氢化蓖麻油脂肪酸组成检测硬脂酸含量偏高,12-羟基硬脂酸偏低为什么呀
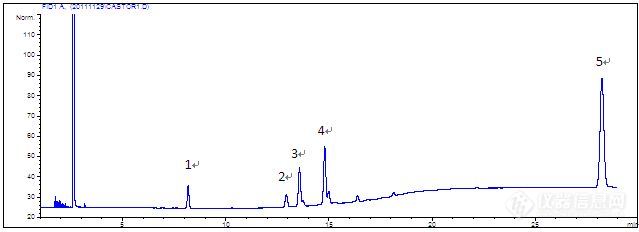
蓖麻油样品 测试项目:含量测定 样品配制: 取蓖麻油40mg,置50mL圆底烧瓶中,加入0.5mol/L氢氧化钠-甲醇溶液5mL,在60℃水浴中回流30min,至油滴全部消失,再加入三氟化硼乙醚-甲醇(1 : 3, v/v) 4mL,回流5min,冷却,精密加入正己烷5mL,振摇5min,分取正己烷,用饱和氯化钠溶液洗涤两次,每次5mL,放置,取上清液,置10mL具塞试管中,加1g 无水硫酸钠脱水,振摇,精密量取上清液1mL,置10mL量瓶中,用正己烷稀释至刻度,摇匀,即得。 仪器型号:Agilent 6890N分析条件 色谱柱: FFAP (柱长为30.0m,内径为0.32mm,膜厚度为0. 5μm) 流动相: 高纯氮 流速: 1.0 mL/min 柱温: 200℃(11min) 5/min 240℃(10min) 检测器: FID 进样量: 1μL 实验谱图http://ng1.17img.cn/bbsfiles/images/2012/07/201207161029_377774_2370618_3.jpg 实验数据 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 1 8.190 186.3 32.4 46742 0.982 2 12.947 160.8 20.5 62736 1.02 3 13.582 478.9 61.9 74025 0.96 4 14.806 672.1[
蓖麻纤维有没有这个名称,标注这个名称检测有没有办法?
实验室做方法转移的时候是用了A和B品牌的对照品,某一次检测时,这两个品牌的都没有了,又用了C品牌的对照品,这样有什么风险吗?更换检测用对照品需要发变更吗?
TLC级别的对照品可以用于液相的检验吗?我们检验用的对照品都是TLC级别的,但现在做出来含量超过100%,可能是对照品的问题?
那位大虾知道蓖麻油的质量指标是什么?请赐教
公司新进的蓖麻油测定时发现按药典方法上面用正己烷5毫升提取一次不能完全提出来,含量只有百分之十几,不知道大家都是怎么操作的[img]https://ng1.17img.cn/bbsfiles/images/2019/12/201912100733389029_7043_3396781_3.png[/img]
2017年的情人节晚上,想必大家已经被一条突如其来的新闻刷屏了——“据韩国媒体报道,朝鲜最高领导人金正恩之兄金正男,在马来西亚机场被暗杀”。尸检结果初步断定致其死亡的化合物为蓖麻毒素或河豚毒素,这两种毒药分别可从蓖麻种子和河豚体内获取,简便易得。关于有毒化合物,你有什么想法?
有人做过2015版中国药典的氢化蓖麻油脂肪酸组成吗?为什么会12羟基硬脂酸甲酯偏低呢?前处理的回流萃取,哪一步需要注意呢?
蓖麻油方法: GC基质: 植物油应用编号: 101924化合物: 1-棕榈酸甲酯2-硬脂酸甲酯3-油酸甲酯4-亚油酸甲酯5-蓖麻油酸甲酯固定相: DM-FFAP色谱柱/前处理小柱: DM-FFAP 30m x 0.32mm x 0.5u商品编号: 7633 样品前处理: 取蓖麻油40mg,置50mL圆底烧瓶中,加入0.5mol/L氢氧化钠-甲醇溶液5mL,在60℃水浴中回流30min,至油滴全部消失,再加入三氟化硼乙醚-甲醇(1 : 3, v/v) 4mL,回流5min,冷却,精密加入正己烷5mL,振摇5min,分取正己烷,用饱和氯化钠溶液洗涤两次,每次5mL,放置,取上清液,置10mL具塞试管中,加1g 无水硫酸钠脱水,振摇,精密量取上清液1mL,置10mL量瓶中,用正己烷稀释至刻度,摇匀,即得。 色谱条件: 色谱柱: FFAP (柱长为30.0m,内径为0.32mm,膜厚度为0. 5μm)流动相: 高纯氮流速: 1.0 mL/min柱温: 200℃(11min) 5/min 240℃(10min)检测器: FID进样量: 1μL 关键字 :蓖麻油;1-棕榈酸甲酯;硬脂酸甲酯;油酸甲酯;-亚油酸甲酯;蓖麻油酸甲酯;gc;DM-FFAPhttp://www.dikma.com.cn/Public/Uploads/images/213(1).JPG峰号保留时间min峰面积μV*s峰高μV理论塔板数NUSP拖尾因子分离度18.190186.332.4467420.982212.947160.820.5627361.02313.582478.961.9740250.96414.806672.191.6945571.06528.3251596180.63126230.98备注说明1- 棕榈酸甲酯2- 硬脂酸甲酯3- 油酸甲酯4- 亚油酸甲酯5-[font='Tim
[font=SimSun, STSong, &]食糖的螨虫检测扩项,需要买对照品吗[/font]
最近在做氢化蓖麻油脂肪酸组成,按药典方法做 ,每次都是12-羟基硬脂酸出峰偏低,感觉没有衍生化完全,各位大神能告知一下有什么注意点吗。我每次做出来其他峰面积都差不多,就是12-羟基硬脂酸的峰面积有差异,而且以面积归一法计含量也只有70%到不了要求的78-91%。
有哪位大神做过2015版中国药典的氢化蓖麻油脂肪酸组成呢?为什么我做出来俩结果不平行 两针对比下来有一针12羟基硬脂酸甲酯偏低 而硬脂酸甲酯偏高 难道二者之间存在转化关系么?还有实验过程中有什么需要注意的吗?
我是做中药检验的,以前用的对照品溶液都经0.45滤膜过滤后使用,还做过某两种对照品溶液的测试,峰面积差异不大。但最近我使用黄芩苷对照品时发现检品含量总是很高,反复测试才找到原因,过滤后对照品的峰面积为:3249725.000,没过滤的对照品峰面积为:4933873.000,差异很大,不知道各位朋友使用对照品溶液时是否也经过0.45滤膜过滤?有没有文件规定对照品溶液能否过滤?
大家好,请问一下,你们在做液相时的对照品,必须要干燥24小时吗?如果对照品上介绍只放室温,没有说在配时,要干燥24小时,可以不干燥吗?
食品微生物检测实验室必须要有单独的阳性对照室吗?检测项目:菌落总数、大肠、霉菌和酵母菌。
曾与昨晚在本版发出求助帖,求助蓖麻籽油的检测方法 [url]http://bbs.instrument.com.cn/shtml/20101009/2850465/[/url] ,非常感谢核桃(pjs123)版主的及时应助!同时也很惊讶于核桃版主快速解决问题的能力——怎么能那么快就搜索到相应标准了呢?赞啊赞~~仔细看了一下核桃版主发上来的标准,发现里面还有不少配套标准我没有。继续求助哦!需要求助的标准有:1、 GB/T5524 2、 GB/T5525 3、 GB/T55264、 GB/T55275、 GB/T55286、 GB/T55297、 GB/T55308、 GB/T55329、 GB/T553410、GB/T668211、GB/T22460
最近小弟按照欧盟的标准做氢化蓖麻油的镍的限度,限度是不超过1ppm,可是厂商的报告是0.1ppm,并且我做的结果老是忽高忽低,第一天测的是1.2ppm,第二天测得是1.3ppm,第三天测得1.5ppm,第四天测得是0.15ppm,同时第四天的样品空白值比前几天高很多,也测了以前合格的样品,可检测结果又高出了限度值。很是不解。我是用石墨炉法,微波消解!请教各位大侠,在配制、消解和测量的的过程中,要注意哪些细节啊?非常感谢!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP