木糖醇、山梨醇、甘露醇标准品那里可以买到?如何用液相分离?
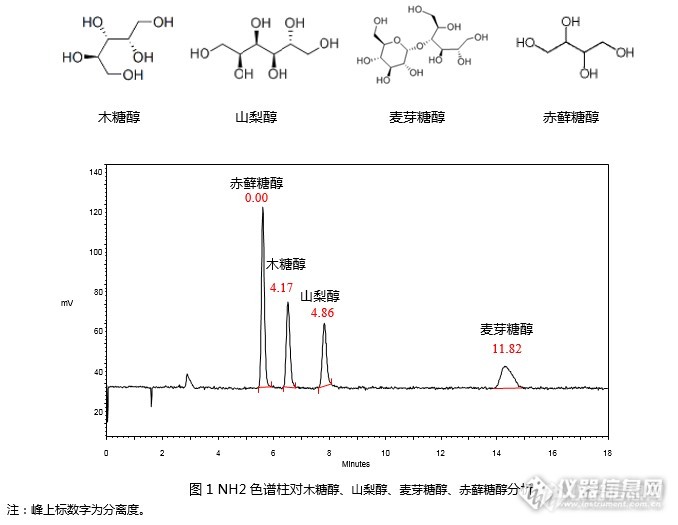
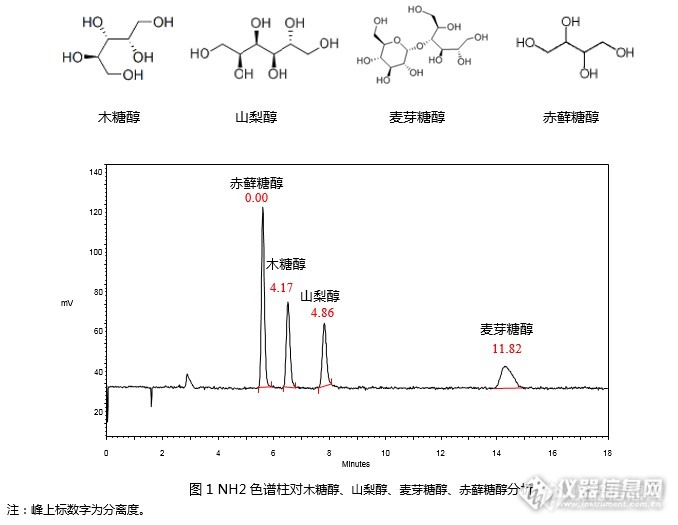
[align=center][b][color=black]GB 5009.279-2016 [/color][color=black]食品安全国家标准[/color][color=black] [/color][color=black]食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定([/color][color=black]RI[/color][color=black])[/color][/b][/align][align=center][b][color=black][/color][/b][/align][align=left][/align][color=black][/color][align=left][color=black]本实验根据《[/color][color=black]GB 5009.279-2016 [/color][color=black]食品安全国家标准[/color][color=black] [/color][color=black]食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定》第一法,使用示差折光检测法[/color]对[color=black]木糖醇、山梨醇、麦芽糖醇、赤藓糖醇[/color][color=black]4[/color][color=black]种[/color]标准品进行分析,并对标准曲线进行考察。[/align][align=center][/align][align=center][img=,600,163]http://ng1.17img.cn/bbsfiles/images/2017/08/201708100910_01_2222981_3.png[/img][/align][align=left]使用资生堂氨基柱CAPCELL PAK NH2 UG80 S5 4.6 mm i.d. [color=black]× [/color]250 mm(GQAD 05507)依据国标方法进行分析,可以实现4种糖醇的良好分析(见图1)。 [/align][align=center][/align][align=center][img=,690,336]http://ng1.17img.cn/bbsfiles/images/2017/08/201708100911_01_2222981_3.png[/img][/align][align=center][/align][align=center][/align][align=left]进一步,依据标准,配制1.6 mg/mL, 2.4 mg/mL, 3.2 mg/mL, 4.0 mg/mL, 4.8 mg/mL, 6.0 mg/mL系列标准工作液,以峰面积为纵坐标,标准工作液浓度为横坐标,绘制标准曲线。[/align][align=left]如图2~5,[color=black]赤藓糖醇、木糖醇、山梨醇、麦芽糖醇[/color]在1.6mg/mL~6.0 mg/mL浓度范围内线性良好,相关系数R[sup]2[/sup]均在0.999以上。[/align][align=center][/align][align=center][img=,582,328]http://ng1.17img.cn/bbsfiles/images/2017/08/201708100911_03_2222981_3.png[/img][img=,547,322]http://ng1.17img.cn/bbsfiles/images/2017/08/201708100912_01_2222981_3.png[/img][/align][align=center][img=,563,326]http://ng1.17img.cn/bbsfiles/images/2017/08/201708100913_01_2222981_3.png[/img][/align][align=center][img=,560,320]http://ng1.17img.cn/bbsfiles/images/2017/08/201708100917_01_2222981_3.png[/img][/align][align=center][/align]综上,依据《[color=black]GB 5009.279-2016 [/color][color=black]食品安全国家标准[/color][color=black] [/color][color=black]食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定》第一法,[/color]使用资生堂CAPCELL PAK NH2 UG80 S5 4.6 mm i.d. × 250 mm(GQAD 05507)色谱柱,以示差折光检测器进行检测,对[color=black]木糖醇、山梨醇、麦芽糖醇和赤藓糖醇[/color]标准品能够得到良好分析结果。在1.6 mg/mL~6.0 mg/mL浓度范围内绘制标准曲线,相关系数R[sup]2[/sup]均在0.999以上,能够得到良好线性关系。[align=center][/align]注: 图中色谱峰线条不平滑是由于图像在复制过程中解像度问题引起的。
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网[em0805]
[align=center]GB 5009.279-2016 食品安全国家标准 食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定-NQAD[/align]《GB 5009.279-2016 食品安全国家标准食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定》第二法中推荐使用蒸发光散射检测器对4种糖醇进行检测。本实验室使用资生堂高灵敏度气溶胶通用型检测器NQAD对该项目进行检测。使用资生堂氨基柱CAPCELL PAK NH2 UG80 S5 4.6 mm i.d. × 250 mm(GQAD 05507)依据国标方法进行分析,可以实现4种糖醇的良好分析(见图1)。[img=,678,525]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030937_01_2222981_3.png[/img][img=,611,257]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030937_02_2222981_3.png[/img]进一步对标准曲线进行绘制,依据国家标准,以峰面积为纵坐标,标准工作液浓度为横坐标,以赤藓糖醇浓度为0.14 mg/mL, 0.21 mg/mL, 0.28 mg/mL, 0.35 mg/mL, 0.42 mg/mL, 0.49 mg/mL,木糖醇、山梨醇、麦芽糖醇浓度为0.10 mg/mL, 0.15 mg/mL, 0.20 mg/mL, 0.25 mg/mL, 0.30 mg/mL, 0.35 mg/mL的混合系列标准工作液,进行标准曲线绘制。如图2~5所示,赤藓糖醇在0.14 mg/mL~0.49 mg/mL浓度范围内,木糖醇、山梨醇、麦芽糖醇在0.1 mg/mL~0.35 mg/mL浓度范围内线性良好,相关系数R[sup]2[/sup]均在0.99以上。[img=,534,330]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_01_2222981_3.png[/img][img=,573,327]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_02_2222981_3.png[/img][img=,573,326]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_03_2222981_3.png[/img][img=,556,342]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_04_2222981_3.png[/img]注:图中色谱峰线条不平滑是由于图像在复制过程中解像度问题引起的。
我想做液相,需要葡萄糖酸标准品,各位有做过的吗,给个建议。谢谢。另外,测酒中的糖份用液相的话,一般用什么柱子。我听说NH2柱,行吗?
谁购买过D-木糖,L-阿拉伯糖,D-半乳糖,D-甘露糖的标准品用来做高效液相,价格是多少?进口的价格比较高,国产的纯度怎么样呀?谢谢大家
食品安全国家标准 食品营养强化剂 葡萄糖酸亚铁
食品安全国家标准 食品营养强化剂 葡萄糖酸铜
食品安全国家标准 食品营养强化剂 葡萄糖酸锰
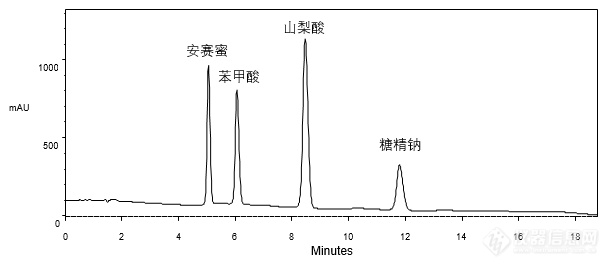
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
2011年 第8号 根据《中华人民共和国食品安全法》、卫生部等9部门《关于加强食品添加剂监督管理工作的通知》(卫监督发〔2009〕89号)和卫生部2011年第6号公告等规定,我部组织中国疾病预防控制中心参照国际标准,指定D-甘露糖醇等58个食品添加剂产品标准。 特此公告。 附件: 1.D-甘露糖醇等58个食品添加剂产品标准目录(见下文) 2.D-甘露糖醇等58个食品添加剂产品标准 二○一一年三月十八日
GB 26404-2011 食品安全国家标准 食品添加剂 赤藓糖醇

问题一:山苯糖国标中有计算标准溶液配置时三种标物的称样量,但是计算后发现它的浓度并不是1000mg/L,而是远大于该浓度,为什么称样量会比计算的理论值大?与纯度有关系?就算是纯度原因,国标中也不会这样直接把称样量多少写出来吧。比如GB 5009.35-2016食品合成着色剂的测定:准确称取按其纯度折算为100%质量的柠檬黄......各0.1g......配成水溶液1.00mg/ml。问题二:另一个问题是糖精钠烘干,想请教各位老师是用什么装着烘干的?是否可以用进样瓶装着烘?如果用称量瓶的话,感觉会污染标品啊,直接把标物品放进去烘可以吗?[img=,690,516]https://ng1.17img.cn/bbsfiles/images/2022/04/202204021130306462_6385_3438623_3.jpg!w690x516.jpg[/img]
GB 26404-2011 食品安全国家标准 食品添加剂 赤藓糖醇
各位大神,我用DNS分光法测糖蜜中还原糖,网上没买到相对应的质控样,只买到了95%的D-葡萄糖标准品,我打算用分析纯无水葡萄糖做标准曲线,用95%的D-葡萄糖标准品做准确度验证,因为样品的还原糖高达48%,不打算做加标回收率,如何把95%的D葡萄糖标准品配制成10mg/ml的标准溶液做准确度验证?
在做国标 GB5009. 28 — 2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定对照品配制疑惑 标准溶液配制:苯甲酸、山梨酸和糖精钠(以糖精计)标准储备溶液( 1000mg / L ):分别准确称取苯甲酸钠、山梨酸钾和糖精钠 0.118g 、 0. 134g 和 0.117g (精确到 0.0001g ),用水溶解并分别定容至 100mL 。于 4℃贮存,保存期为 6 个月。当使用苯甲酸和山梨酸标准品时,需要用甲醇溶解并定容。红色这句话的意思是想用适量的甲醇溶解,然后在用水定容,还是用甲醇定容,感觉怪怪的?有那位老师可以指导一下, 共享一下经验,谢谢。
各有关单位: 根据《食品安全法》及其实施条例的规定,我委组织拟订了《食品安全国家标准食品营养强化剂 葡萄糖酸镁》等9项食品安全国家标准(征求意见稿),现向社会公开征求意见。请于2016年3月30日前登录食品安全国家标准管理信息系统(http://bz.cfsa.net.cn/cfsa_aiguo)在线提交反馈意见。 附件: 1.《食品安全国家标准 食品营养强化剂 葡萄糖酸镁》(征求意见稿)及编制说明 2.《食品安全国家标准 食品营养强化剂 5’单磷酸胞苷》(征求意见稿)及编制说明 3.《食品安全国家标准 食品营养强化剂 醋酸视黄酯(醋酸维生素A)》(征求意见稿)及编制说明 4.《食品安全国家标准 食品营养强化剂 D-泛酸钠》(征求意见稿)及编制说明 5.《食品安全国家标准 食品营养强化剂 多聚果糖》(征求意见稿)及编制说明 6.《食品安全国家标准 食品营养强化剂 低聚果糖》(征求意见稿)及编制说明 7.《食品安全国家标准 食品营养强化剂 氯化锌》(征求意见稿)及编制说明 8.《食品安全国家标准 食品营养强化剂 乙酸锌》(征求意见稿)及编制说明 9.《食品安全国家标准 食品营养强化剂 海藻碘》(征求意见稿)及编制说明
各位大神,我用DNS分光法测糖蜜中还原糖,网上没买到相对应的质控样,只买到了95%的D-葡萄糖标准品,我打算用分析纯无水葡萄糖做标准曲线,用95%的D-葡萄糖标准品做准确度验证,因为样品的还原糖高达48%,不打算做加标回收率,如何把95%的D葡萄糖标准品配制成10mg/ml的标准溶液做准确度验证?
卫监督食便函〔2011〕4号 各有关单位:根据《食品安全法》及其实施条例的规定,按照卫生部等9部门《关于加强食品添加剂监督管理工作的通知》(卫监督发〔2009〕89号)的要求,拟指定D-甘露糖醇等58个食品添加剂标准。现公开征求意见,请于2011年1月14日前按下列方式反馈意见:传真010-67711813或电子信箱gb2760@gmail.com。附件:D-甘露糖醇等58个食品添加剂.rar 二○一一年一月五日
各位大神,我用DNS分光法测糖蜜中还原糖,网上没买到相对应的质控样,只买到了95%的D-葡萄糖标准品,我打算用分析纯无水葡萄糖做标准曲线,用95%的D-葡萄糖标准品做准确度验证,因为样品的还原糖高达48%,不打算做加标回收率,如何把95%的D葡萄糖标准品配制成10mg/ml的标准溶液做准确度验证?
β-Glucuronidase/aryl sulfatase β-葡萄糖醛酸酶\芳香基硫酸酯酶( 葡萄糖苷酸酶/硫酸芳酯酶 ) 标准品——检瘦肉精等用的除了北京希凯创新科技有限公司提供,还可以从哪里购买么?
[color=#444444]GB 5009.28-2016中是这么写的:苯甲酸、山梨酸和糖精钠(以糖精计)标准储备溶液(1000mg/L):分别准确称取苯甲酸钠、山梨酸钾和糖精钠[/color][color=#444444][b]0.118g、0.134g和0.117g[/b][/color][color=#444444](精确到0.0001g),用水溶解并分别定容至100mL。于4℃贮存,保存期为6个月。当使用苯甲酸和山梨酸标准品时,需要用甲醇溶解并定容。注:糖精钠含结晶水,使用前需在120 ℃烘4h,干燥器中冷却至室温后备用。[/color][color=#444444]苯甲酸钠好理解,苯甲酸分子量122,苯甲酸钠分子量144。0.118*122/144=0.1[/color][color=#444444]但是无水糖精钠分子量为205,二水糖精钠分子量为241,糖精分子量为183,如果算[/color][color=#444444]糖精[/color][color=#444444]的话应该是:205/183=1.12,称0.1的话应该是0.112,为什么是0.117呢?[/color][color=#444444]如果算的是二水糖精钠的话,应该是0.1*205/241=0.085(GB 5009.28-2003《食品中糖精钠的测定》中称的就是0.0851g),所以2016版的国标为什么要称0.117g,求大神解答一下。。。[/color]
恶喹酸标准品用什么溶解啊?我们用的甲醇,都没溶!看网上说加氢氧化钠,但是以后怎么用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测呢?
恶喹酸标准品用什么溶解啊?我们用的甲醇,都没溶!如果用氢氧化钠助溶还能用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测吗?
国家药监局有个检测13种降糖类药物的标准,包括格列本脲、丁二胍等等,绝大多数组分是微溶或难溶于甲醇,基本不溶于水。而标准是用甲醇来溶解。有人做过吗,标准品的溶解是如何解决的?
近日,《食品中胆固醇的测定高效液相色谱法》等五项国家标准审定会在中国计量科学研究院举行。该5项标准将有望解决我国食品中胆固醇、糖、糖醇、脂肪和膳食纤维含量检测缺乏标准的问题。 这5项标准分别是《食品中胆固醇的测定高效液相色谱法》、《食品中总脂肪、饱和脂肪酸、不饱和脂肪酸的测定水解提取-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法》、《食品中果糖、葡萄糖、蔗糖、麦芽糖、乳糖的测定高效液相色谱法》、《食品中木糖醇、山梨醇、麦芽糖醇的测定高效液相色谱法》和《含抗性麦芽糊精食品中总膳食纤维的测定酶重量法- 液相色谱法》。这5项标准是在大量的研究工作基础上制定的,有力解决了我国食品中胆固醇、糖、糖醇、脂肪和膳食纤维含量检测缺乏标准的问题,填补了国内空白,经检测机构证实切实可行,为这些食品营养成分和标签标示成分的检测提供了技术依据,对于构建我国食品营养成分的检测体系具有重要的现实意义。同时,标准的制定立足我国实际,充分考虑与国际接轨,对食品进出口贸易也将起到积极的推动作用,有力支持我国食品标签工作的实施。
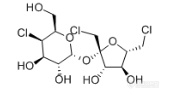
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-NQAD检测器[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种甜味剂具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011004470313_2453_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]本实验按照[b]《GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定》[/b]方法,使用[b][color=#ff0000]高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD)[/color][/b]对三氯蔗糖标准品进行了分析。色谱柱选择中等极性普适型[color=#3333ff][b]CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 150 mm[/b][/color],得到结果如图1所示。三氯蔗糖保留时间为12.709min,[b]与标准谱图保留时间基本一致,理论塔板数为9992,不对称因子为1.06,峰形良好。[/b][align=center][b][img=,690,497]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006155125_1559_2222981_3.png!w690x497.jpg[/img][/b][/align][align=center]图1 三氯蔗糖标准品分析色谱图[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[b][img=,633,176]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006367633_3986_2222981_3.png!w633x176.jpg[/img]附图:GB方法中标准色谱图[/b][align=center][b][img=,690,448]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011007162573_9264_2222981_3.png!w690x448.jpg[/img][/b][/align][b][/b]接下来,按照国标要求配制三氯蔗糖工作液,浓度分别为0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。[b][color=#3333ff]由于NQAD检测器原理与常规蒸发光散射检测器ELSD不同,能够直接得到线性回归结果,不需要做对数方程,更加简单快捷。[/color][/b]线性结果如图2所示,R[sup]2[/sup]=0.996,得到良好线性结果。同时,我们根据标准曲线最低浓度的信噪比计算出定量限(以S/N=10计)约为3 μg/mL,[b][color=#ff0000]能够实现三氯蔗糖的高灵敏度检出[/color][/b]。[align=center][img=,658,399]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011008425185_5014_2222981_3.png!w658x399.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-RI[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种[url=http://baike.sogou.com/v130009.htm][color=windowtext]甜味剂[/color][/url]具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080920210187_4197_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]实验室前期按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD),得到了三氯蔗糖标准品的良好分析结果。本实验按照相同条件,使用示差折光检测器(RI)对三氯蔗糖标准品进行分析。色谱柱同样选择中等极性的普适型色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 150 mm,得到结果如图1所示。三氯蔗糖保留时间为12.400min,与标准谱图保留时间基本一致,理论塔板数为12350,不对称因子为0.95,峰形良好。[align=center][img=,690,489]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945469257_8172_2222981_3.png!w690x489.jpg[/img][/align][align=center]图1 三氯蔗糖标准品分析色谱图(0.4 mg/mL)[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[img=,472,187]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945471937_6640_2222981_3.png!w472x187.jpg[/img][align=center][img=,690,435]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080946205953_7240_2222981_3.png!w690x435.jpg[/img][/align][align=center]附图:GB方法中标准色谱图[/align]接下来,按照国标要求配制三氯蔗糖工作液,0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。线性实验结果如图2所示,R[sup]2[/sup]=0.9939,得到良好线性结果。同时,由于低浓度0.02 mg/mL、0.05 mg/ mL标准品溶液均未检出色谱峰,因此根据标准曲线最高浓度的信噪比计算出检出限(以S/N=3计)约为0.17 mg/ mL。[align=center][img=,650,398]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080947051037_4812_2222981_3.png!w650x398.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]综上,按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用示差检测器(RI)进行检测,以及CAPCELL PAK C[sub]18[/sub] MGII S5 4.6 mm i.d. ×150 mm色谱柱进行分析,可得到三氯蔗糖标准品的良好线性分析结果;但RI检测器的检测灵敏度较低。
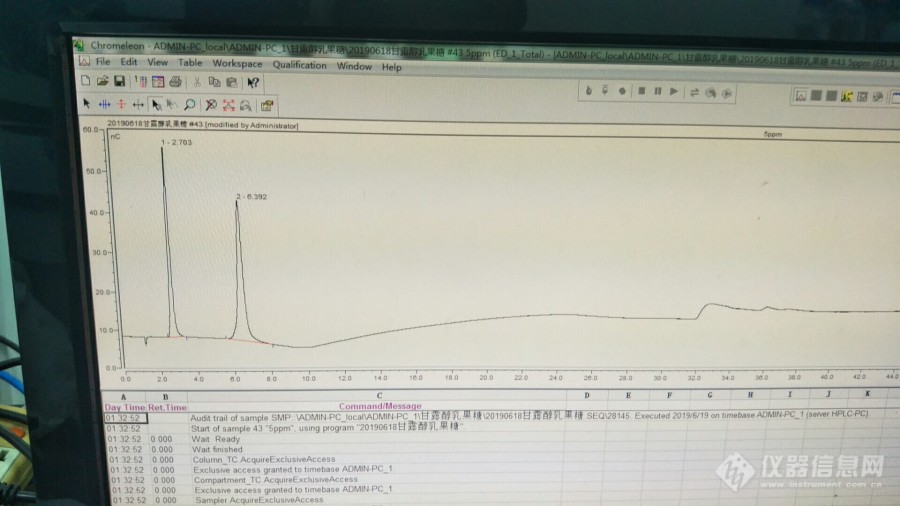
糖标准品的困惑-从购买到假乳果糖谈起最近为了测定一批甘露醇和乳果糖的样品(采用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法),原来乳果糖对照品几乎用完了,需要重新购,于是我在学校的虫洞采购平台上订购,从麦克林公司购买了乳果糖的对照品,一般而言,我不可能去验证糖对照品的准确性。将对照品给学生后,就把以前的色谱条件交给她们,让她们去做。做了几次后,学生反映,跟原来的结果不一样,保留时间对不上,我自己也没仔细查看,就跟学生说,这样品做了数百个了,条件和色谱柱都是固定不变了,不会有问题。不行,继续试,不行,再继续做。在几次失败后,学生无意用了原来剩下的标样试了一下,发现跟新买来的标样保留不一样,跑过来跟我说,这时我才有点醒悟。在仔细观察了色谱图,并查阅色谱条件后,我明白学生为什么做不出来的原因。买来的乳果糖标样不对,因为原来的乳果糖标样不止买过一次,十年来都没出过问题。而且从色谱图的保留时间看,买来的乳果糖标样出峰时间在单糖的位置,肯定是糖标样有问题。本来我想狠狠批评学生,但也怪我,我之前没有仔细查看色谱图,我负有不可推卸的责任!学生第一次做,缺乏经验,但对于单糖双糖的保留时间位置,应该有感性的认识,我只能教育他们,并告诉他们判断的依据。客户在催,我只能再从网上订购,这次另外换了一家公司,从迈瑞尔公司订购了乳果糖。试剂到后,让学生先测试一下,是否是乳果糖。实验的结果跟从麦克林买来一样,不对,还是单糖不是双糖(乳果糖是双糖)。[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/08/201908192347296833_4583_1617661_3.jpg!w690x387.jpg[/img]甘露醇和购买的假乳果糖的[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]图[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/08/201908192348130616_5401_1617661_3.jpg!w690x387.jpg[/img]甘露醇和原来的乳果糖(原来试剂时间放长了,有变质,图不是很好,但能说明问题,)二次从二个不同的厂家采购相同的乳果糖,结果表明二者都不是乳果糖,都是单糖,可能是半乳糖(没核对),这个漏洞也太大了吧,说明麦克林和迈瑞尔对进货的乳果糖都没有检验,或者检验有缺失,万一出错,这是多么可怕的事,会对实验造成严重的后果,如果一个第一次使用这种糖的人,如果缺乏判断经验,不可想象。我不敢再买了,那个是真的?那些小的试剂公司更不敢碰。用户一边在催实验结果,另一边试剂还没着落,买进口保险吗?或许如此,但价格高很多了。时间上也不见得来得及。为了保险起见,我再次联系了第三家试剂公司,阿拉丁,通过电话沟通,让他们确认试剂的可靠性,如果有检验的话,单糖和双糖是极容易区分的。在他们保证后,第三次购买了乳果糖,实验结果表明,这次购买的是正确的,跟留下来的乳果糖是同一种糖,实验终于可以进行了。通过这次事件,我预感到,试剂市场可能存在一定的乱象,一旦出现滥竽充数,如果不加检验,一些小公司可能根本没有能力判断。错误试剂会对实验结果造成严重的后果。但是让用户去检验试剂的可靠性,虽然我能做到,但是测试成本会远远高于购买的成本。而一般用户,根本没有能力。这也许是低价造成的恶果!大家是否有相同的经历!!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP