1.从引物自身着手,重新设计引物,这是最根本解决这一问题的办法。2.可能模板有问题,模板浓度过小,适当加大模板量。3.Taq酶,引物,Mg2+浓度可能过高,可降低它们的浓度。4.将上下引物混合后,在100℃的沸水中煮5分钟,然后迅速拿出至于冰块之上瞬时冷却,这时再加入反应体系当中,引物二聚体就会消失的。理由:引物可能会发生发夹结构,自身环化等结构,在100℃的沸水中煮5分钟可使引物变为单链,以减少二聚体。不过有人认为在PCR仪上95度变性5min也同样达到目的,而且成功试过通过延长退火时间也可以消除引物二聚体。5.所配MIX中加5%的甘油或者5%的DMSO,可以增强特异性。6.PCR反应体系的配制在冰上进行,最后加Taq酶,PCR结束后,产物勿放置在室温下过长时间,有人认为室温下有些Taq酶会将多余的引物合成为二聚体。7.增加循环数。8.降低退火温度后有条带,则应逐渐提高温度,若提高温度的同时产物量减少,则考虑增加Mg2+浓度(根据扩增片断长度而定,片段长则相应镁离子浓度应该高一些)。9.若降低退火温度,发现还是只有引物二聚体,而且镁离子的浓度在20-25mmol/l没有区别,则考虑Buffer等试剂没有完全融解、混匀,导致吸取的试剂浓度不对。10.以上次的PCR产物作模板二次PCR,可以提高引物与模板的特异性,减少引物二聚体,如果两次时间间隔短的话,可以把原产物稀释100-1000倍,如果间隔较长可以稀释50-100倍。
甲基环戊二烯二聚体有没有异构体?在[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]上出几个峰?用什么色谱柱?
附件是原料巯基丙酮用酒精稀释后进的gcms,请问巯基丙酮二聚体的峰到底是14.866还是22.072,或者说两者都是?还有,根据香料通则,这个东西的含量要达到95%,根据图上看有个很大的巯基丙酮,含量应该不到95%,巯基丙酮是本来就有的呢还是二聚体分解出来的?大家做原料控制的时候怎么做的呢?
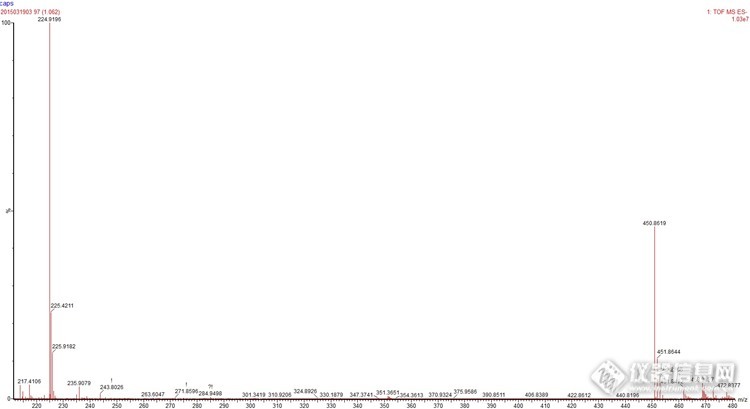
ES-做的,224.9同位素相差是0.5,450.9同位素相差1,这个是二聚体吗?分子量是452?大神解释下http://ng1.17img.cn/bbsfiles/images/2015/03/201503201055_538947_2359430_3.jpg
如题,乙偶因二聚体在气相上能出得来不?如何测定乙偶姻中二聚体的含量?
求二聚酸中单体、二聚体、三聚体含量测定方法?
在解析ESI低分辨时,如何区分有二倍关系的是二具体,还是因为出现了多点带电,比如说,我打了个ESI+低分辨300-500有个离子峰M/Z=415.2,响应强度为2.40e3。而600-1000范围有两个较强的离子峰M/Z=785.4,M/Z=807.4,响应强度为681。从785.4和807.4可判断出807.4为加Na,785.4为加H,415.2也是加Na。那么分子量到底是多少,按二聚体算的话,分子量应该为392,。如果说是415.2带了两个电荷,那么分子量是不是应该就是784。(请高手给讲讲,这ESI源打质谱如何判断分子量)图上传不上
我的化合物是属于含金属锂离子的复合物,然后又加了溴离子,(即得到了这样的二聚体,其他实验已证实该二聚体的存在),做了高分辨质谱发现了双电荷的峰,这个双电荷的 峰正好是显示含2个锂离子的二聚体的分子量那么我能不能判断我的东西在加了溴离子之后可以形成二聚体注:我的化合物不能络合两个金属锂离子,不加溴离子之前的高分辨质谱中也没发现二聚体的峰
请各位老师谈谈方向,我想试试GC测三氧化硫以及二聚体三聚体的含量。
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]如何检测乙偶姻二聚体?查沸点279,出峰却很早,与乙偶姻出峰在一个位置上。
我想从猪肝中分离纯化金属硫蛋白,样品经过Sephadex G-75凝胶柱后,提出其中MT二聚体的蛋白峰,分子量14000左右,经过浓缩后上弱阴离子交换柱,采用0.01-1.0M醋酸铵缓冲液线性梯度洗脱,PH8.3,但是经过一次实验后溶液的PH降低,大量洗脱平衡后仍不能达到8.3,现在我该怎么办?请赐教.
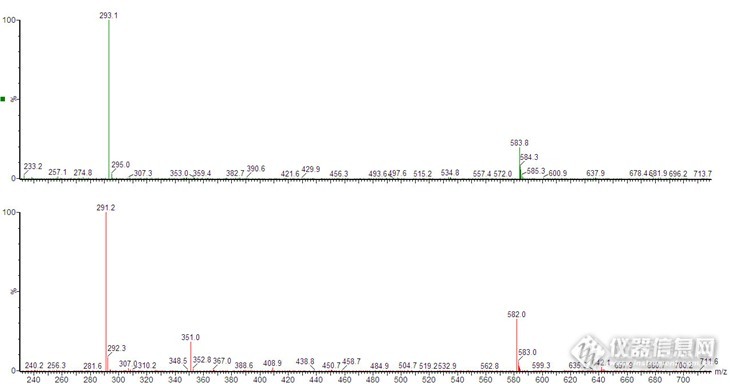
我有一个质谱图,已知分子离子峰在不同模式下分别为291和293,那么二聚体峰如何解释呢?见附图。data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAABPwAAAKfCAYAAAD6jdY4AAAgAElEQVR4AezdPVLb2v84/uP/ZAtQ3CY1A8VtswAYuuyBCRuAJt3nf7vbkA3AZA+3y8AC0n4LGGqaFGYR/kmyjy3JkiwbA/LhxUyip/P4OrIs3uhhNMl+gh8CBAgQIECAAAECBAgQIECAAAECBJIQ+P+S6IVOECBAgAABAgQIECBAgAABAgQIECBQCAj42REIECBAgAABAgQIECBAgAABAgQIJCQg4JfQYOoKAQIECBAgQIAAAQIECBAgQIAAAQE/+wABAgQIECBAgAABAgQIECBAgACBhAQE/BIaTF0hQIAAAQIECBAgQIAAAQIECBAgIOBnHyBAgAABAgQIECBAgAABAgQIECCQkICAX0KDqSsECBAgQIAAAQIECBAgQIAAAQIEBPzsAwQIECBAgAABAgQIECBAgAABAgQSEhDwS2gwdYUAAQIECBAgQIAAAQIECBAgQICAgJ99gAABAgQIECBAgAABAgQIECBAgEBCAgJ+CQ2mrhAgQIAAAQIECBAgQIAAAQIECBAQ8LMPECBAgAABAgQIECBAgAABAgQIEEhIQMAvocHUFQIECBAgQIAAAQIECBAgQIAAAQICfvYBAgQIECBAgAABAgQIECBAgAABAgkJCPglNJi6QoAAAQIECBAgQIAAAQIECBAgQEDAzz5AgAABAgQIECBAgAABAgQIECBAICEBAb+EBlNXCBAgQIAAAQIECBAgQIAAAQIECAj42QcIECBAgAABAgQIECBAgAABAgQIJCQg4JfQYOoKAQIECBAgQIAAAQIECBAgQIAAAQE/+wABAgQIECBAgAABAgQIECBAgACBhAQE/BIaTF0hQIAAAQIECBAgQIAAAQIECBAgIOBnHyBAgAABAgQIECBAgAABAgQIECCQkICAX0KD+SG78nwXLkejMJr9u3koKZS3Xd6F59Km8HAzzzMaXYa7ysZywtJ8Ud5NKFdR2mqWAAECBAgQIECAAAECBAgQIDAIAQG/QQyDRmwm8BBu9k/Cwf0kTCbZv/vrcH4Ug3fTbeF2XGwbn/4K+zEamAfujh6zTTHfQTjZXxXIm5b3Y7OGykWAAAECBAgQIECAAAECBAgQeDMBAb83o1bR1gUefofzi9vw9XBW8uHXcHvxIzyNs+XnP+ExXIez471i497xWbg+/zm9km/vOFxNrsJsUwiHX7KUj+FPy1V+z3eX2dWAR+Hx+jpcbL0TCiRAgAABAgQIECBAgAABAgQIbFdAwG+7nkp7S4HDb2FydRymIb284nF42uASvOe7n1ng8DT8vSio2ou/zsI4u4Lw6uvn6npLBAgQIECAAAECBAgQIECAAIEBCgj4DXBQNGkzgWngbnbF395f4SCch9+zB+4V28Ls6r9Y/OwZf9ldweH2ezlwGBNMp3uHh6WgYnWbJQIECBAgQIAAAQIECBAgQIDA0AQE/IY2ItqzkUB+2+3+yUG4n1/xdxi+jW/D49H0hR7/hrPsdt9a0cWtvfmz//o8w6+W1yIBAgQIECBAgAABAgQIECBAYKACAn4DHRjN6i8wDfZlV+mNv4X4OL8idwzo5bfjHofsdt+L8Hm/odwVz/BryGEVAQIECBAgQIAAAQIECBAgQGCwAp8G2zINI9BDYH5l36QW7KvnLV7icRC+tD2nr57eMgECBAgQIECAAAECBAgQIEBgRwV6XOH3HO4uR+Fm9iy0RT+n60ej6S2Tl3f1V5yu2r4oyRyBjQSyZ/D9mz9/r35lX1HYQ7gZXU7fypstP/x3En5cf5leAfhwk7119ybMd+k8GNj10o6NGicTAQIECBAgQIAAAQIECBAgQOB9BFYE/PKg3X44WXrz6Wz9wX2YZLdLTib34eBkPyyCfqu2v09n1ZqWQBHEy17EcbI/DTrH4PM0OJ09w694Nt9029HjbRh/m93wm73dd3z7GI5mwerR/lM4mz/7LwsO3oxK+3JaZnpDgAABAgQIECBAgAABAgQIpC8wygJ2k6ZuTm+VzCJ9F9fhOnvbaTibhBgvCfnbTfd/hdPxVTiOt0jmV039/BzGeeBk1famCq0jQIAAAQIECBAgQIAAAQIECBAgQODFAh1X+J2G+/zqvauv4XO9mvFT+FG/BXL/c7j48RTGedpV2+vlWSZAgAABAgQIECBAgAABAgQIECBAYCsCrQG/vePj6htPS9U9/3ksLZVnH8Of7FF+q7aXc5gnQIAAAQIECBAgQIAAAQIECBAgQGB7Aq0Bv+1V0VxSfN5a07Q5h7UEtisw+me03QKVRoAAAQIECBAgQIAAAQIECBAYgMCnTdqw99dBS7aD8Ff2TL9V2/PMTY8OzIN/fggQIECAAAECBAgQIECAAAECBAgQ2Fxgsyv8ys/ri3UXz+37HPbz5VXbYx5TAgQIECBAgAABAgQIECBAgAABAgS2KrBZwG/v73B6cR6Obh5mjXkIN0fn4eL071C8tHfV9q12QWEECBAgQIAAAQIECBAgQIAAAQIECESBzQJ+WVjv+Gocbh+PwvQZfEfh8XYcro6LcF9W9qrtsXpTAgQIECBAgAABAgQIECBAgAABAgS2KTDKnqU32WaBLykrPsNvQE16SXfkHbhA/tKOyf8Gs/sPXEvzCBAgQIAAAQIECBAgQIAAgV0R2PAKv13pnnYSIECAAAECBAgQIECAAAECBAgQ+FgCAn4fa7z1lgABAgQIECBAgAABAgQIECBAIHEBAb/EB1j3CBAgQIAAAQIECBAgQIAAAQIEPpaAgN/HGm+9JUCAAAECBAgQIECAAAECBAgQSFxAwC/xAdY9AgQIECBAgAABAgQIECBAgACBjyUg4PexxltvCRAgQIAAAQIECBAgQIAAAQIEEhcQ8Et8gHWPAAECBAgQIECAAAECBAgQIEDgYwkI+H2s8dZbAgQIECBAgAABAgQIECBAgACBxAUE/BIfYN0jQIAAAQIECBAgQIAAAQIECBD4WAICfh9rvPWWAAECBAgQIECAAAECBAgQIEAgcQEBv8QHWPcIECBAgAABAgQIECBAgAABAgQ+loCA38cab70lQIAAAQIECBAgQIAAAQIECBBIXEDAL/EB1j0CBAgQIECAAAECBAgQIECAAIGPJSDg97HGW28JECBAgAABAgQIECBAgAABAgQSFxDwS3yAdY8AAQIECBAgQIAAAQIECBAgQOBjCQj4fazx1lsCBAgQIECAAAECBAgQIECAAIHEBQT8Eh9g3SNAgAABAgQIECBAgAABAgQIEPhYAgJ+H2u89ZYAAQIECBAgQIAAAQIECBAgQCBxAQG/xAdY9wgQIECAAAECBAgQIECAAAECBD6WgIDfxxpvvSVAgAABAgQIECBAgAABAgQIEEhcQMAv8QHWPQIECBAgQIAAAQIECBAgQIAAgY8lIOD3scZbbwkQIECAAAECBAgQIECAAAECBBIX+DSE/o1GoyE0QxsIECBAgAABAgQIECBAgAABAgQI7LzAIK7wm0wmIf/nhwABAgQIECBAgAABAgQIECBAgACBlwkMIuD3si7ITYAAAQIECBAgQIAAAQIECBAgQIBAFBDwixKmBAgQIECAAAECBAgQIECAAAECBBIQEPBLYBB1gQABAgQIECBAgAABAgQIECBAgEAUEPCLEqYECBAgQIAAAQIECBAgQIAAAQIEEhAQ8EtgEHWBAAECBAgQIECAAAECBAgQIECAQBQQ8IsSpgQIECBAgAABAgQIECBAgAABAgQSEBDwS2AQdYEAAQIECBAgQIAAAQIECBAgQIBAFBDwixKmBAgQIECAAAECBAgQIECAAAECBBIQEPBLYBB1gQABAgQIECBAgAABAgQIECBAgEAUEPCLEqYECBAgQIAAAQIECBAgQIAAAQIEEhAQ8EtgEHWBA
我正在制备双特异抗体,采用半分子互换方法,即一半A 一半B,但是交联后,仍有少量的A和B的污染,因为我是想得到需要的双特异性抗体,因此需要纯化,请问如何才能做到? 据说可以用分析型CIEX实现 不知道具体的方法和原理如何 望指教,谢谢 我的联系方式时 13936179062 微信 电子邮件是 13936179062@139.com
多方求助无果,希望有高人指点。急急我们实验室在做重组人复合α干扰素(cIFN)的聚合与降解研究。cIFN单体中有两条分子内二硫键,它们在巯基乙醇作用下可以被还原打开,并且在空气中会形成分子间二硫键,进而引起cIFN聚合。再向这些聚合体中添加过量巯基乙醇后绝大多数的聚合体都被离解成单体,但是通过还原SDS-PAGE仍可以看到有微量的二聚体。理论上还原条件下二硫键是不存在的,所以我们推测这个二聚体应该是其他化学键引起的。我想知道的是:1、蛋白质除了二硫键是否还有其他的化学键可以引起蛋白质共价聚合体。2、用什么方法可以鉴定蛋白质聚合体中单体间的化学键?[em0812][em0812][em0811][em0811][em0811]
我是学超分子化学的,以前我用NMR稀释的方法,通过记录特征H化学位移的变化,应用Origin进行非线性拟和可以求得氢键二聚体的结合常数。但对于结合常数高的二聚体,由于NMR灵敏度的限制,在浓度很小的情况下难以进行测定。现打算采用UV-vis(适宜低浓度)来进行测定,但我对UV-vis很陌生,特向大家请教!
岛津HPLC(型号10A)在测定白蛋白多聚体的时应新药典要求进行系统适应性试验之分离度的计算class-LC10工作站中提供了直接计算的功能,但此项功能多数情况下不能给出二聚体峰与单体峰之间的分离度R,观察多张图谱得出只要是两峰中任一峰无理论塔板数则R无数值显示,即使是两峰在图谱上可明显观察到已分离,不明白为什么会出现此情况。PS:测定白蛋白成品时分析时间为70min,在单体峰(28min)之后会出一乙酰色氨酸峰(50min)而此两峰间R可达16左右。成品之前有一组分Ⅴ(生产中间体)样品也会测定其中多聚体含量,因其内不含乙酰色氨酸,故分析时间为50min,奇怪的就是恰恰因为这个原因使得每次单体峰与二聚体峰均可达到药典要求(R2.5)其实组分Ⅴ与成品组成几乎相同,仅仅只是纯度不及后者本人实在是不明白既然R是有公式计算得的,有峰存在,即有公式中的参数值即使是R很下,但也该有个数值吧!请高手帮忙解决一下,不胜感激!~
人血白蛋白多聚体测定法本法系用分子排阻色谱法测定人血白蛋白多聚体含量。照分子排阻色谱法(附录Ⅲ D)测定。色谱条件与系统适用性试验 用亲水硅胶高效体积排阻色谱柱(SEC,排阻极限300kD,粒度10μm),柱直径7.5mm,长60cm;以含1%异丙醇的pH7.0 0.2mol/L磷酸盐缓冲液为流动相;检测波长为280nm ;流速为每分钟0.6ml。取每1ml含蛋白质为12mg 的人血白蛋白溶液20μl,注入色谱柱,记录色谱图,人血白蛋白单体峰与二聚体峰间的分离度应大于1.5,拖尾因子按人血白蛋白单体峰计算应为0.95~1.40。测定法 取供试品适量,用流动相稀释成每1ml约含蛋白质12mg的溶液,取20μl,注入色谱柱,记录色谱图60分钟(色谱柱长60cm)。按面积归一法计算,色谱图中未保留(全排阻)峰的含量(%)除以2,即为人血白蛋白多聚体含量请教:为何除以2呢?很奇怪呀
在做二羟基丙酮的硅烷化分析的时候,出现一个问题:当标准样品的量比较少的时候比如10mg左右的时候,气相上出来的是二羟基丙酮的硅烷化的峰,但是当标品的来那个超过20mg的时候会出来二羟基丙酮硅烷化峰以及二羟基丙酮二聚体硅烷化峰,不知哪位专家做过此物质的分析,可否指点一下。
川西合耳菊(叶格兴嘎保、雨布星嘎布)为常用藏药,来源于菊科合耳菊属植物川西合耳菊Synotis solidaginea (Hand. -Mazz.) C. Jeffrey et Y. L. Chen的干燥地上部分,主要分布于西藏、四川、青海等地区;具有清解毒热、愈伤消炎的功效,常用于治疗跌打损伤、蛇虫毒咬等外伤[1]。川西合耳菊的药用历史悠久,最早记载于《度母本草》,用于治疗疮伤病[2]。藏药经典著作《晶珠本草》记载叶格兴(川西合耳菊)为接骨愈伤药[3]。《四川省藏药材标准》(2020年版)和《西藏自治区地方药材(饮片)质量标准》(2023年版)均收载了川西合耳菊[4-5]。尽管川西合耳菊的植物资源丰富,但是对其化学成分研究报道较少。杨爱梅等[6-7]从川西合耳菊石油醚部位分离鉴定了6个艾里莫芬烷倍半萜类成分。李定祥等[8-9]从川西合耳菊正丁醇部位分离鉴定了16个化合物。本课题组前期在测定四川、西藏、青海等不同产地川西合耳菊中8种黄酮类和酚酸类成分含量的过程中,发现川西合耳菊含有多种结构类型的化学成分[10]。为了阐明川西合耳菊的药效物质基础,开发利用其资源,并为临床应用提供依据,本研究对西藏采集的川西合耳菊地上部位进行了系统的化学成分研究,从醋酸乙酯萃取部位分离鉴定了18个化合物(图1),分别鉴定为(2R,4S,5R,8R,10S)-2-angeloyloxy-8,10-dihydroxy-eremophil-7(11)-en-8,12-olide(1)、(1R,4S,5S,6S,8R,10S)-1,10-epoxy-6,8- dihydroxy-eremophil-7(11)-en-8,12-olide(2)、(4S, 5R,8S,10S)-8,10-dihydroxy-eremophil-7(11)-en-8,12-olide(3)、(4S,5R,8R,10S)-10-hydroxy-eremophil- 7(11)-en-8,12-olide(4)、橐吾内酯[(4S,5R)-eremophil- 1(10),7(11),8(9)-trien-8,12-olide,5]、4,5-O-二咖啡酰奎宁酸甲酯 [4,5-di-O-caffeoylquinic acid methyl ester,6]、异绿原酸C(4,5-di-ca?eoylquinic acid,7)、3,5-O-二咖啡酰基奎宁酸甲酯(3,5-di-O- caffeoylquinic acid methyl ester,8)、异绿原酸A (3,5-di-caffeoylquinic acid,9)、1,4-O-二咖啡酰奎宁酸甲酯(1,4-di-O-caffeoylquinic acid methyl ester,10)、绿原酸(chlorogenic acid,11)、咖啡酰苹果酸 [(2'R)-phaselic acid,12]、咖啡酸甲酯(caffeic acid methyl ester,13)、反式咖啡酸(trans caffeic acid,14)、山柰酚-3-O-(6''-O-乙酰基)-β-D-吡喃葡萄糖苷[kaempferol-3-O-(6''-O-acetyl)-β-D-glucopyranoside,15]、紫云英苷(astragalin,16)、2-苯乙基-β-D-吡喃葡萄糖苷(2-phenylethyl-β-D-glucopyranoside,17)、桦褐孔菌二糖(inotodisaccharide,18)。其中化合物1为新的艾里莫芬烷倍半萜,命名为川西合耳菊内酯A(synotisolidolide A),化合物2、4、5、10、12、13、15、17、18为首次从合耳菊属植物中分离得到。 图片 1 仪器与材料 Bruker Avance Neo 600 MHz核磁共振波谱仪(德国Bruker公司)、UPLC-Q-Orbitrap高分辨质谱仪(美国Thermo Fisher Scientific公司)、Chirascan qCD圆二色光谱仪(英国应用光物理公司)、PuriMAster-3000A制备[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url](上海科哲生化科技有限公司)、聚酰胺(80~120目,浙江省台州市路桥四甲生化塑料厂)。柱色谱硅胶(200~300目)和GF254薄层硅胶板由青岛海洋化工有限公司生产。Ultimate-XB色谱柱 C18(250 mm×10 mm,5 μm)、Ultimate-XB C18反相材料(40~70 μm)、色谱纯甲醇和乙腈均由月旭科技(上海)股份有限公司生产。超纯水由PALL Cascada纯水机(美谷富迪生物仪器有限公司)制备。 川西合耳菊地上部分采自西藏林芝市米瑞乡玉荣增村,经成都中医药大学药学院吕光华教授鉴定为菊科植物川西合耳菊S. solidaginea (Hand. -Mazz.) C. Jeffrey et Y. L. Chen,标本(CXHER20191125)保存于成都中医药大学药学院标本中心。 2 提取与分离 川西合耳菊干燥地上部位9.5 kg,粉碎,用70%乙醇回流提取3次,每次1.5 h。提取液滤过、减压浓缩至稠膏后,将其分散在水中,依次用石油醚、醋酸乙酯和正丁醇萃取。醋酸乙酯萃取部分浓缩至浸膏(399 g),经硅胶柱色谱分离,以二氯甲烷-甲醇(20∶1~0∶1)梯度洗脱,经薄层色谱检测合并后,得到4个流分(Fr. A~D)。 Fr. A(43 g)经硅胶柱色谱,二氯甲烷-甲醇(100∶1~0∶1)梯度洗脱,合并得6个流分(Fr. A1~A6)。Fr. A1(5 g)经硅胶柱色谱分离,石油醚- 二氯甲烷(2∶1~0∶1)梯度洗脱,结晶,得化合物5(382 mg)。Fr. A5(10 g)经硅胶柱色谱,二氯甲烷-甲醇(20∶1~5∶1)梯度洗脱,再经制备高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]纯化,乙腈-水(36∶64~90∶10)梯度洗脱,得化合物1(4.1 mg,tR=66 min)、3(7.1 mg,tR=33 min)和4(3.8 mg,tR=45 min)。 Fr. B(68 g)经硅胶柱色谱分离,二氯甲烷-甲醇(100∶1~0∶1)梯度洗脱,得12个流分(Fr. B1~Fr. B12)。Fr. B2(5 g)经硅胶柱色谱分离、醋酸乙酯-甲醇(20∶1~10∶1)梯度洗脱,制备高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]纯化、甲醇-水(40∶60~65∶35)梯度洗脱,得化合物2(7.1 mg,tR=28 min)。Fr. B4(0.15 g)经制备高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]纯化,甲醇-水(10∶90~55∶45)梯度洗脱,得到化合物14(9.1 mg,tR=27.5 min)。Fr. B7(4 g)经硅胶柱色谱分离,二氯甲烷-甲醇(20∶1~0∶1)梯度洗脱,制备高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]纯化(甲醇-水20∶80~55∶45),得化合物10(6.8 mg,tR=30 min)、15(6.1 mg,tR=38 min)和17(6 mg,tR=26 min)。Fr. B12(0.2 g)经制备高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分离,甲醇-水(20∶80~90∶10)梯度洗脱,得化合物18(22.8 mg,tR=33 min)。 Fr. D(138.6 g)经硅胶柱色谱,二氯甲烷-甲醇(20∶1~0∶1)梯度洗脱,得5个流分(Fr. D1~D5)。Fr. D2(15 g)经聚酰胺柱色谱分离,乙醇-水(10∶90~100∶0)洗脱;C18反相硅胶柱色谱分离,甲醇-水(30∶70~90∶10)洗脱,得Fr. D2.1~D2.4。Fr. D2.1(5 g)经硅胶柱色谱分离,醋酸乙酯-甲醇-水(30∶1∶1~15∶1∶1)梯度洗脱,得化合物11(115.3 mg)、12(872 mg)和13(8 mg)。Fr. D2.2(7 g)经硅胶柱色谱分离,醋酸乙酯-甲醇-水(100∶1∶1~15∶1∶1)洗脱,得化合物6(1.146 2 g)、8(263 mg)和16(392.6 mg)。Fr. D2.4(300 mg)经过制备高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]纯化,甲醇-水(40∶60~70∶30)梯度洗脱,得化合物9(20.4 mg,tR=14 min)和7(10.4 mg,tR=20 min)。 3 结构鉴定 化合物1:白色粉末。(nm):285.14(0.22)。(cm?1): 3 419、1 660、1 614、1 585、1 391、1 354。根据HR-ESI-MS数据([M-H]? m/z 363.181 30,计算值363.181 55),确定其分子式为C20H28O6,不饱和度为7。1H-NMR谱(表1)显示化合物1具有5个甲基信号,其中1个角甲基质子信号δH 1.08 (s, 3H)、3个烯碳甲基信号δH 1.93 (dq, J = 7.2, 1.8 Hz, 3H), 1.84 (brs, 3H), 1.83 (brs, 3H)、1个仲碳甲基信号δH 0.94 (d, J = 6.6 Hz, 3H);1个三取代烯氢质子信号δH 6.09 (qq, J = 7.2, 1.8 Hz, 1H),1个连氧次甲基信号δH 4.84 (m, 1H)。13C-NMR谱(表1)显示该化合物有21个碳信号,包括2个酯羰基信号δC 173.9和168.9,1个三取代双键和1个四取代双键δC 159.7、138.6、129.2和124.0,3个连氧碳信号δC 104.2、75.1和70.2,5个甲基信号δC 20.7、16.5、15.9、15.2和8.4。以上NMR数据表明该化合物中存在1个当归酰氧基。化合物1的NMR数据与已报道的化合物1β-angeloyloxy- 8β,10β-dihydroxy-eremophil-7(11)-en-8α,12-olide的相似[11]。两者的第1个区别在于已知化合物中连接在C-1位的当归酰氧基在化合物1中连接在C-2位,1H-1H COSY谱(图2)中的自旋偶合片段H3-15/H-4/H2-3/H-2/H2-1和HMBC谱(图2)中H-2与C-1'相关,证实了以上区别;第2个区别在于两者在C-8位的化学位移不同,已知化合物C-8位的化学位移从δC 101.5向低场位移至δC 104.2,提示化合物1在C-8上的羟基构型与已知化合物的相反,为α构型。比较化合物1与1β-angeloyloxy-8β,10β- dihydroxy-eremophil-7(11)-en-8α,12-olide在C-4、C-5和C-10的化学位移(δC 46.8 vs45.8,32.5 vs33.4,75.1 vs75.7),表明两者在C-4、C-5和C-10的相对构型一致,均为β构型。根据菊科植物中艾里莫酚烷倍半萜的生源规律,一般C-4和C-5上的甲基均为β构型[12]。该化合物的NOESY中观察到H-2/H-4相关,说明H-2为α构型。综上所述,化合物1具有2种异构体1a (2R,4S,5R,8R,10S) 和1b (2S,4R,5S, 8S,10R)。通过比较实验与计算ECD谱(图3),发现该化合物的实验ECD曲线与1a (2R,4S,5R,8R,10S) 的曲线完全吻合。因此,化合物1的结构鉴定为 (2R,4S,5R,8R,10S)-2-angeloyloxy- 8,10-dihydroxy-eremophil-7(11)-en-8,12-olide。经Sci- Finder检索,化合物1为新化合物,命名为川西合耳菊内酯A(synotisolidolide A)。 图片 图片 图片 化合物2:白色粉末。分子式为C15H20O5。1H-NMR (600 MHz, CD3OD) δ: 4.84 (1H, d, J = 1.8 Hz, H-6), 3.13 (1H, d, J= 4.8 Hz, H-1), 2.26 (1H, d, J = 13.2 Hz, H-9a), 2.05 (1H, m, H-4), 2.02 (3H, brs, H-13), 1.94 (1H, m, H-2a), 1.92 (1H, m, H-2b), 1.71 (1H, m, H-3a), 1.63 (1H, d, J = 13.2 Hz, H-9b), 1.35 (1H, m, H-3b), 1.05 (3H, d, J = 7.2 Hz, H-15), 1.00 (3H, s, H-14);13C-NMR (150 MHz, CD3OD) δ: 174.3 (C-12), 161.7 (C-7), 125.0 (C-11), 103.0 (C-8), 72.9 (C-6), 63.8 (C-1), 62.8 (C-10), 45.4 (C-5), 44.6 (C-9), 32.8 (C-4), 24.7 (C-3), 21.3 (C-2), 16.4 (C-15), 14.0 (C-14), 8.7 (C-13)。以上数据与文献报道一致[13],故鉴定化合物2为 (1R,4S,5S,6S,8R,10S)-1,10-epoxy- 6,8-dihydroxy-eremophil-7(11)-en-8,12-olide。 化合物3:白色粉末。分子式为C15H22O4。1H-NMR (600MHz, CD3OD) δ: 2.68 (1H, d, J = 14.0 Hz, H-6a), 2.46 (1H, d, J = 14.0 Hz, H-6b), 2.37 (1H, d, J = 14.4 Hz, H-9a), 2.00 (1H, d, J = 14.4 Hz, H-9b), 1.82 (3H, brs, H-13), 1.76 (1H, m, H-1a), 1.58 (1H, m, H-2b), 1.40 (4H, m, H-1b, 2a, 3b, 4a), 1.31 (1H, td, J= 13.6, 4.6 Hz, H-3a), 1.04 (3H, s, H-14), 0.87 (3H, d, J = 6.0 Hz, H-15);13C-NMR (150 MHz, CD3OD) δ: 174.1 (C-12), 160.5 (C-7), 123.5 (C-11), 104.9 (C-8), 75.2 (C-10), 47.4 (C-5), 43.9 (C-9), 35.7 (C-1), 34.6 (C-4), 31.4 (C-6), 30.9 (C-4), 23.0 (C-2), 16.8 (C-15), 15.2 (C-14), 8.4 (C-13)。以上数据与文献报道一致[14],故鉴定化合物3为 (4S,5R,8S,10S)-8,10-dihydroxy- eremophil-7(11)-en-8,12-olide。 化合物4:白色粉末。分子式为C15H22O3。1H-NMR (600 MHz, CD3OD) δ:5.02 (1H, t, J = 9.0 Hz, H-8), 2.74 (1H, d, J = 13.8 Hz, H-6a), 2.47 (1H, d, J = 13.8 Hz, H-6b), 2.12 (1H, dd, J = 12.6, 6.6 Hz, H-9a), 1.97 (1H, dd, J = 12.6, 10.8 Hz, H-9b), 1.81 (1H, dd, J = 3.0, 1.2 Hz, H-1a), 1.79 (3H, brs, H-13), 1.57 (1H, m, H-2a), 1.46 (1H, m, H-1b), 1.45 (1H, m, H-4), 1.42 (1H, m, H-3a), 1.41 (1H, m, H-2b), 1.30 (1H, m, H-3b), 1.02 (3H, s, H-14), 0.86 (3H, d, J = 6.6 Hz, H-15);13C-NMR (150 MHz, CD3OD) δ: 177.5 (C-12), 164.4 (C-7), 121.1 (C-11), 80.8 (C-8), 75.4 (C-10), 46.2 (C-5), 42.1 (C-9), 36.6 (C-1), 34.8 (C-4), 32.7 (C-6), 31.1 (C-3), 23.3 (C-2), 16.6 (C-15), 15.2 (C-14), 8.3 (C-13)。以上数据与文献报道一致[15],故鉴定化合物4为 (4S,5R,8R,10S)-10-hydroxy- eremophil-7(11)-en-8,12-olide。 化合物5:黄色柱状结晶(甲醇)。分子式为C15H18O2。1H-NMR (600 MHz, CDCl3) δ: 5.94 (1H, s, H-9), 5.80 (1H, t, J = 3.6 Hz, H-1), 2.84 (1H, d, J = 16.2 Hz, H-6a), 2.25 (2H, m, H-2), 2.23 (1H, m, H-6b), 1.91 (3H, brs, H-13), 1.74 (1H, m, H-4), 1.57 (2H, m, H-3), 1.00 (3H, d, J = 6.6 Hz, H-15), 0.97 (3H, s, H-14);13C-NMR (150 MHz, CDCl3) δ: 171.7 (C-12), 147.6 (C-8), 147.4 (C-7), 139.3 (C-10), 131.4 (C-1), 120.6 (C-11), 109.7 (C-9), 39.0 (C-4), 37.8 (C-5), 34.9 (C-6), 26.6 (C-3), 26.9 (C-2), 19.7 (C-14), 15.8 (C-15), 8.6 (C-13)。以上数据与文献报道一致[16],故鉴定化合物5为橐吾内酯。 化合物6:淡黄色粉末。分子式为C26H26O12。1H-NMR (600 MHz, CD3OD) δ: 7.60 (1H, d, J = 15.6 Hz, H-7''), 7.51 (1H, d, J = 15.6 Hz, H-7'), 7.03 (1H, d, J = 1.8 Hz, H-2''), 7.02 (1H, d, J = 1.8 Hz, H-2'), 6.93 (1H, d, J = 8.4 Hz, H-6''), 6.92 (1H, d, J = 8.4 Hz, H-6'), 6.76 (2H, d, J = 8.4 Hz, H-5', 5''), 6.30 (1H, d, J = 15.6 Hz, H-8''), 6.17 (1H, d, J = 15.6 Hz, H-8'), 5.55 (1H, m, H-5), 5.12 (1H, dd, J = 8.4, 3.0 Hz, H-4), 4.35 (1H, m, H-3), 3.72 (3H, s, 8-OCH3), 2.32 (1H, m, H-6a), 2.25 (2H, m, H-2a), 2.09 (1H, m, H-6b);13C-NMR (150 MHz, CD3OD) δ: 175.2 (C=O, C-7), 168.5 (C-1''), 167.9 (C-1'), 149.7 (C-7'), 149.7 (C-7''), 147.7 (C-3''), 147.7 (C-3'), 146.8 (C-6''), 146.7 (C-6'), 127.7 (C-4''), 127.5 (C-4'), 123.2 (C-9'), 123.2 (C-9''), 116.5 (C-8''), 116.5 (C-8'), 115.2 (C-5''), 115.1 (C-5'), 114.7 (C-2''), 114.5 (C-2'), 75.8 (C-4), 74.9 (C-1), 69.1 (C-5), 68.6 (C-3), 53.1 (-OCH3, C-8), 38.5 (C-2), 38.3 (C-6)。以上数据与文献报道一致[17],故鉴定化合物6为4,5-O-二咖啡酰奎宁酸甲酯。 化合物7:白色粉末。分子式为C25H24O12。1H-NMR (600 MHz, CD3OD) δ: 7.60 (1H, d, J = 15.6 Hz, H-7'), 7.52 (1H, d, J = 15.6 Hz, H-7''), 7.03 (1H, brs, H-2''), 7.00 (1H, brs, H-2'), 6.92 (1H, d, J = 8.4 Hz, H-6''), 6.90 (1H, d, J = 8.4 Hz, H-6'), 6.75 (1H, d, J = 8.4 Hz, H-5''), 6.74 (1H, d, J = 8.4 Hz, H-5'), 6.29 (1H, d, J = 15.6 Hz, H-8''), 6.19 (1H, d, J = 15.6 Hz, H-8'), 5.62 (1H, m, H-5), 5.12 ( 1H, dd, J = 9.0, 3.0 Hz, H-4), 4.37 (1H, m, H-3), 2.30 (1H, m, H-6a), 2.28 (1H, m, H-2a), 2.24 (1H, m, H-6b), 2.11 (1H, m, H-2b);13C-NMR (150 MHz, CD3OD)δ: 176.9 (C-7), 168.6 (C-9'), 168.2 (C-9''), 149.7 (C-4', 4''), 147.7 (C-7'), 147.6 (C-7''), 146.8 (C-3'), 146.7 (C-3''), 127.7 (C-1'), 127.6 (C-1''), 123.1 (C-6'), 123.1 (C-6''), 116.5 (C-5'), 116.5 (C-5''), 115.2 (C-2'), 115.1 (C-2''), 114.7 (C-8'), 114.7 (C-8''), 76.1 (C-1), 75.7 (C-4), 69.3 (C-5), 69.0 (C-3), 39.3 (C-6), 38.4 (C-2)。以上数据与文献报道一致[18],故鉴定化合物7为异绿原酸C。 化合物8:白色粉末。分子式为C26H26O12。1H-NMR (600 MHz, CD3OD) δ: 7.62 (1H, d, J = 15.6 Hz, H-7''), 7.55 (1H, d, J = 15.6 Hz, H-7'), 7.07 (2H, s, H-2', 2''), 6.97 (2H, d, J = 8.4 Hz, H-6', 6''), 6.80 (1H, d, J = 8
1.O-H伸缩振动羧酸在浓溶液或固态中因强氢键成二聚体。 强氢键源于离子共振,阻碍游离羟基振动,仅稀非极性溶剂或蒸气相中可见(约3520 cm?1)。 二聚体O-H振动宽且强,范围3300~2500 cm?1,常集中于3000 cm?1,伴弱C-H振动。 长波长精细结构为倍频与复合频。 β-二酮等也有此吸收,但较弱,C-O振动频率较低。与醚类溶剂形成分子间氢键,O-H吸收约3100 cm?1。 2.C-O伸缩振动羧酸C-O振动强于酮,单体约1760 cm?1。二聚体对称,仅不对称振动有吸收,氢键与共振降低频率至1720~1706 cm?1。 分子内氢键影响更大,如水杨酸1665 cm?1,对羟基苯甲酸1680 cm?1。 不饱和共轭轻微降低频率,α,β-不饱和及芳基共轭酸二聚体约1710~1680 cm?1。 α位电负性取代基(如卤素)轻微增加频率,旋转异构致双重谱带。 3.C-O伸缩与O-H弯曲振动羧酸红外光谱中,C-O伸缩约13201210 cm?1,O-H弯曲约14401395 cm?1,两者有相互作用。 二聚体C-O伸缩强吸收约1315~1280 cm?1,长链脂肪酸呈双峰。 O-H面外弯曲特征谱带约920 cm?1,中等强度峰宽。
跟大家分享一下,聚氨酯的IR分析 v3250-3500 ms OH伸缩振动、NHCO的顺式NH伸缩振动。 v2940、2860 s CH2、CH3伸缩振动。 v2240-2280 s NCO特征吸收峰。 v2120 s 碳化二亚胺吸收峰。 v1770-1785 s 脲二酮环(二聚体)中的C=O。 v1715-1750 vs 酯基C=O、酰胺I键C=O。 v1689-1710 s 异腈脲酸酯(三聚体)中C=O(1408-1430也有峰) v1600-1615 苯环C=C骨架伸缩振动。 v1520-1560 ms 酰胺II键(N-H)变形振动。 v1450-1470 CH2变形振动、CH3非对称变形振动。 v1380 CH3对称变形振动 v1225-1235 聚酯C-O伸缩或OH变形振动 v1060-1150 宽s C-O-C(脂肪族醚)吸收峰。
http://ng1.17img.cn/bbsfiles/images/2016/01/201601032253_580610_1621538_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601032253_580611_1621538_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601032253_580612_1621538_3.jpg帮一个同学问的:自己合成的样品,用液相色谱法分离,总是分不开。一个大峰,峰形不太好看,件图1.色谱柱:sugar-Ca;色谱柱要求见图2。分析标准品可以分开,见图3.现在流动相为水和乙醇,流速和柱温都调整了,还是分不开,请大家指点。
高聚物摩尔质量不仅反映了高聚物分子的大小,而且直接关系到它的物理性能,是个重要的基本参数。与一般的无机物或低分子的有机物不同,高聚物多是摩尔质量大小不同的大分子混合物,所以通常所测高聚物摩尔质量是一个统计平均值。测定高聚物摩尔质量的方法很多,而不同方法所得平均摩尔质量也有所不同。比较起来,黏度法设备简单,操作方便,并有很好的实验精度,是常用的方法之一。用该法求得的摩尔质量称为黏均摩尔质量。黏度法测高聚物溶液摩尔质量时.恒温槽1套;乌贝路德黏度计1支;分析天平1台;移液管(10mL,2支、5mL,1支);停表1只;洗耳球1个;橡皮管夹2个;橡皮管(约5cm长,2根);吊锤1个。聚丙烯酰胺(或聚乙烯醇) ;NaNO3(3mol• dm-3、1mol• dm-3)。外推法求[η]
[align=center][b][font=宋体]附录Ⅵ[/font][font=Times New Roman] Q[/font][font=宋体]人血白蛋白多聚体测定法[/font][/b][/align][font=宋体][size=3]本法系用分子排阻色谱法测定人血白蛋白多聚体含量。[/size][/font][size=3][font=宋体]照分子排阻色谱法(附录Ⅲ[/font][font=Times New Roman] D[/font][font=宋体])测定。[/font][/size][size=3][b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=Times New Roman] [/font][font=宋体]用亲水硅胶高效体积排阻色谱柱([/font][font=Times New Roman]SEC[/font][font=宋体],排阻极限[/font][font=Times New Roman]300kD[/font][font=宋体],粒度[/font][font=Times New Roman]10[/font][font=宋体]μ[/font][font=Times New Roman]m[/font][font=宋体]),柱直径[/font][font=Times New Roman]7[/font][font=宋体].[/font][font=Times New Roman]5mm[/font][font=Times New Roman],[/font][font=宋体]长[/font][font=Times New Roman]60cm[/font][font=宋体];以含[/font][font=Times New Roman]1[/font][font=宋体]%异丙醇的[/font][font=Times New Roman]pH7[/font][font=宋体].[/font][font=Times New Roman]0 0[/font][font=宋体].[/font][font=Times New Roman]2mol/L[/font][font=宋体]磷酸盐缓冲液[取[/font][font=Times New Roman]0[/font][font=宋体].[/font][font=Times New Roman]5mol/L[/font][font=宋体]磷酸二氢钠[/font][font=Times New Roman]200ml[/font][font=宋体]、[/font][font=Times New Roman]0[/font][font=宋体].[/font][font=Times New Roman]5mol/L[/font][font=宋体]磷酸氢二钠[/font][font=Times New Roman]420ml[/font][font=宋体]、异丙醇[/font][font=Times New Roman]15[/font][font=宋体].[/font][font=Times New Roman]5ml[/font][font=宋体]及水[/font][font=Times New Roman]914.5ml[/font][font=宋体],混匀]为流动相;检测波长为[/font][font=Times New Roman]280nm [/font][font=宋体];流速为每分钟[/font][font=Times New Roman]0[/font][font=宋体].[/font][font=Times New Roman]6ml[/font][font=宋体]。取每[/font][font=Times New Roman]1ml[/font][font=宋体]含蛋白质为[/font][font=Times New Roman]12mg [/font][font=宋体]的人血白蛋白溶液[/font][font=Times New Roman]20[/font][font=宋体]μ[/font][font=Times New Roman]l[/font][font=宋体],注入色谱柱,记录色谱图[/font][font=Times New Roman],[/font][font=宋体]人血白蛋白单体峰与二聚体峰间的分离度应大于[/font][font=Times New Roman]1[/font][font=宋体].[/font][font=Times New Roman]5[/font][font=宋体],拖尾因子按人血白蛋白单体峰计算应为[/font][font=Times New Roman]0[/font][font=宋体].[/font][font=Times New Roman]95[/font][font=宋体]~[/font][font=Times New Roman]1[/font][font=宋体].[/font][font=Times New Roman]40[/font][font=宋体]。[/font][/size][size=3][b][font=宋体]测定法[/font][/b][font=Times New Roman] [/font][font=宋体]取供试品适量,用流动相稀释成每[/font][font=Times New Roman]1ml[/font][font=宋体]约含蛋白质[/font][font=Times New Roman]12mg[/font][font=宋体]的溶液[/font][font=Times New Roman],[/font][font=宋体]取[/font][font=Times New Roman]20[/font][font=宋体]μ[/font][font=Times New Roman]l[/font][font=宋体],注入色谱柱,记录色谱图[/font][font=Times New Roman]60[/font][font=宋体]分钟[/font][s][font=宋体](色谱柱长[/font][/s][s][font=Times New Roman]60cm[/font][/s][s][font=宋体])[/font][/s][font=宋体]。[/font][/size][size=3][color=#fe2419][font=宋体]按面积归一法计算,色谱图中未保留(全排阻)峰的含量[/font][font=Times New Roman](%)[/font][font=宋体]除以[/font][font=Times New Roman]2[/font][font=宋体],即为人血白蛋白多聚体含量。[/font][/color][/size][size=3][font=Times New Roman][b][color=#0021b0]计算方法不是很明白,为什么要除以2 ??? 全排阻的峰指的是哪一个??谢谢[/color][/b][/font][/size]
LC—MS分析葡萄糖,ESI(+)离子模式,+(二聚体加氢峰较高),有没有遇到类似情况的呢?一级质谱,即出现很多脱水峰,请问正常吗?
大家好! 有个问题,我做的是亲油性的粉末,可以很好的分散到二氯甲烷中,不能分散到乙醇中。 采用不同的分散剂洗涤并离心,干燥后重新分散到二氯甲烷中做透射电镜分析。所用的分散剂分别为乙醇和二氯甲烷。结果是有一定差异的:乙醇洗的颗粒团聚体周围有很多小颗粒,而二氯甲烷洗的颗粒团聚体周围则相对比较干净。 对原因我实在不能解释,请大家帮我出个主意。 谢谢大家!http://ng1.17img.cn/bbsfiles/images/2011/07/201107011532_302594_2284983_3.gifhttp://ng1.17img.cn/bbsfiles/images/2011/07/201107011533_302596_2284983_3.gif
聚合酶链式反应(Polymerase Chain Reaction,PCR)是体外酶促合成特异DNA片段的一种方法,为最常用的分子生物学技术之一。典型的PCR由(1)高温变性模板;(2)引物与模板退火;(3)引物沿模板延伸三步反应组成一个循环,通过多次循环反应,使目的DNA得以迅速扩增。其主要步骤是:将待扩增的模板DNA置高温下(通常为93℃-94℃)使其变性解成单链;人工合成的两个寡核苷酸引物在其合适的复性温度下分别与目的基因两侧的两条单链互补结合,两个引物在模板上结合的位置决定了扩增片段的长短;耐热的DNA聚合酶(Taq酶)在72℃将单核苷酸从引物的3’端开始掺入,以目的基因为模板从5’→3’方向延伸,合成DNA的新互补链。 PCR能快速特异扩增任何已知目的基因或DNA片段,并能轻易在皮克(pg)水平起始DNA混合物中的目的基因扩增达到纳克、微克、毫克级的特异性DNA片段。因此,PCR技术一经问世就被迅速而广泛地用于分子生物学的各个领域。它不仅可以用于基因的分离、克隆和核苷酸序列分析,还可以用于突变体和重组体的构建,基因表达调控的研究,基因多态性的分析,遗传病和传染病的诊断,肿瘤机制的探索,法医鉴定等诸多方面。通常,PCR在分子克隆和DNA分析中有着以下多种用途:(1) 生成双链DNA中的特异序列作为探针;(2) 由少量mRNA生成 cDNA文库;(3) 从cDNA中克隆某些基因;(4) 生成大量DNA以进行序列测定;(5) 突变的分析;(6) 染色体步移;(7) RAPD、AFLP、RFLP等DNA多态性分析等。一、试剂准备1. DNA模版2.对应目的基因的特异引物3.10×PCR Buffer 4.2mM dNTPmix:含dATP、dCTP、dGTP、dTTP各2mM5.Taq酶二、操作步骤 1.在冰浴中,按以下次序将各成分加入一无菌0.5ml离心管中。 10×PCR buffer 5 μl dNTP mix (2mM) 4 μl 引物1(10pM) 2 μl 引物2(10pM) 2 μl Taq酶 (2U/μl) 1 μl DNA模板(50ng-1μg/μl) 1 μl 加ddH2O至 50 μl 视PCR仪有无热盖,不加或添加石蜡油。2. 调整好反应程序。将上述混合液稍加离心,立即置PCR仪上,执行扩增。一般:在93℃预变性3-5min,进入循环扩增阶段:93℃ 40s → 58℃ 30s → 72℃ 60s,循环30-35次,最后在72℃ 保温7min。3. 结束反应,PCR产物放置于4℃待电泳检测或-20℃长期保存。4.PCR的电泳检测:如在反应管中加有石蜡油,需用100μl氯仿进行抽提反应混合液,以除去石蜡油;否则,直接取5-10μl电泳检测。三、PCR反应体系的组成与反应条件的优化 PCR反应体系由反应缓冲液(10×PCR Buffer)、脱氧核苷三磷酸底物(dNTPmix)、耐热DNA聚合酶(Taq酶)、寡聚核苷酸引物(Primer1,Primer2)、靶序列(DNA模板)五部分组成。各个组份都能影响PCR结果的好坏。1. 反应缓冲液:一般随Taq DNA聚合酶供应。标准缓冲液含:50mM KCl,10mM Tris-HCl(pH8.3室温),1.5mM MgCl2。Mg2+的浓度对反应的特异性及产量有着显著影响。浓度过高,使反应特异性降低;浓度过低,使产物减少。在各种单核苷酸浓度为200μM时,Mg2+为1.5mM较合适。若样品中含EDTA或其它螯合物,可适当增加Mg2+的浓度。在高浓度DNA及dNTP条件下进行反应时,也必须相应调节Mg2+的浓度。据经验,一般以1.5-2mM(终浓度)较好。2. dNTP :高浓度dNTP易产生错误掺入,过高则可能不扩增;但浓度过低,将降低反应产物的产量。PCR中常用终浓度为50-400μM的dNTP。四种脱氧三磷酸核苷酸的浓度应相同,如果其中任何一种的浓度明显不同于其它几种时(偏高或偏低),就会诱发聚合酶的错误掺入作用,降低合成速度,过早终止延伸反应。此外,dNTP能与Mg2+结合,使游离的Mg2+浓度降低。因此,dNTP的浓度直接影响到反应中起重要作用的Mg2+浓度。3. Taq DNA聚合酶酶:在100μl反应体系中,一般加入2-4U的酶量,足以达到每min延伸1000-4000个核苷酸的掺入速度。酶量过多将导致产生非特异性产物。但是,不同的公司或不同批次的产品常有很大的差异,由于酶的浓度对PCR反应影响极大,因此应当作预试验或使用厂家推荐的浓度。当降低反应体积时(如20μl或50μl),一般酶的用量仍不小于2U,否则反应效率将降低。4. 引物:引物是决定PCR结果的关键,引物设计在PCR反应中极为重要。要保证PCR反应能准确、特异、有效地对模板DNA进行扩增,通常引物设计要遵循以下几条原则:⑴ 引物的长度以15-30bp为宜,一般(G+C)的含量在45-55%,Tm值高于55℃。应尽量避免数个嘌呤或嘧啶的连续排列,碱基的分布应表现出是随机的。⑵ 引物的3’端不应与引物内部有互补,避免引物内部形成二级结构,两个引物在3’端不应出现同源性,以免形成引物二聚体。3’端末位碱基在很大程度上影响着Taq酶的延伸效率。两条引物间配对碱基数少于5个,引物自身配对若形成茎环结构,茎的碱基对数不能超过3个由于影响引物设计的因素比较多,现常常利用计算机辅助设计。⑶ 人工合成的寡聚核苷酸引物需经PAGE或离子交换HPLC进行纯化。⑷ 引物浓度不宜偏高,浓度过高有两个弊端:一是容易形成引物二聚体(primer-dimer),二是当扩增微量靶序列并且起始材料又比较粗时,容易产生非特异性产物。一般说来,用低浓度引物不仅经济,而且反应特异性也较好。一般用0.25-0.5pM/μl较好。⑸ 引物一般用TE配制成较高浓度的母液(约100μM),保存于-20℃。使用前取出其中一部分用ddH2O配制成10μM或20μM的工作液。5. 模板:PCR对模板的要求不高,单、双链DNA均可作为PCR的样品。虽然PCR可以用极微量的样品(甚至是来自单一细胞的DNA)作为摸板,但为了保证反应的特异性,一般还宜用μg水平的基因组DNA或104拷贝的待扩增片段作为起始材料。原材料可以是粗制品,某些材料甚至仅需用溶剂一步提取之后即可用于扩增,但混有任何蛋白酶、核酸酶、Taq DNA聚合酶抑制剂以及能结合DNA的蛋白,将可能干扰PCR反应。6. PCR循环加快,即相对减少变性、复性、延伸的时间,可增加产物的特异性。四、注意事项1.PCR反应应该在一个没有DNA污染的干净环境中进行。最好设立一个专用的PCR实验室。2.纯化模板所选用的方法对污染的风险有极大影响。一般而言,只要能够得到可靠的结果,纯化的方法越简单越好。3.所有试剂都应该没有核酸和核酸酶的污染。操作过程中均应戴手套。4.PCR试剂配制应使用最高质量的新鲜双蒸水,采用0.22μm滤膜过滤除菌或高压灭菌。5.试剂都应该以大体积配制,试验一下是否满意,然后分装成仅够一次使用的量储存,从而确保实验与实验之间的连续性。6.试剂或样品准备过程中都要使用一次性灭菌的塑料瓶和管子,玻璃器皿应洗涤干净并高压灭菌。7.PCR的样品应在冰浴上化开,并且要充分混匀。
溶液中,大分子分子量达到多少就不能用核磁共振的方法来测了?二聚体或者寡聚体也算作一个分子吗?还有是不是NMR仪器的兆数越大可以测的生物大分子的分子量上限就会越大?谢谢!
在检测多聚体含量的时候,跟半个月前做的样品检测的单体峰的峰高以及峰面积都都有明显的下降,但是含量的比例还是保持稳定的。单体峰的峰高之前可以达到350-500mV都有的,基本稳定在400mV左右,但是今天做出来的单体峰的峰高却只有160mV,而且多聚体和二聚体也是一样的降了近一半,还有流动相是一样的,都是磷酸盐,柱子是去年7月份买的,请问下这个原因可能有哪些方面造成的?
PCR引物设计的原则 引物设计有3 条基本原则:首先引物与模板的序列要紧密互补,其次引物与引物之间避免形成稳定的二聚体或发夹结构,再次引物不能在模板的非目的位点引发DNA 聚合反应(即错配)。 具体实现这3 条基本原则需要考虑到诸多因素,如引物长度(primer length),产物长度(product length),序列Tm 值(melting temperature),引物与模板形成双链的内部稳定性(internal stability, 用∆G 值反映),形成引物二聚体(primer dimer)及发夹结构(duplex formation and hairpin)的能值,在错配位点(false priming site)的引发效率,引物及产物的GC 含量(composition),等等。必要时还需对引物进行修饰,如增加限制性内切酶位点,引进突变等。根据有关参考资料和笔者在实践中的总结,引物设计应注意如下要点: 1. 引物的长度一般为15-30 bp,常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。 2. 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错配。引物3’端出现3 个以上的连续碱基,如GGG 或CCC,也会使错误引发机率增加3. 引物3’端的末位碱基对Taq 酶的DNA 合成效率有较大的影响。不同的末位碱基在错配位置导致不同的扩增效率,末位碱基为A 的错配效率明显高于其他3 个碱基,因此应当避免在引物的3’端使用碱基A。另外,引物二聚体或发夹结构也可能导致PCR 反应失败。5’端序列对PCR 影响不太大,因此常用来引进修饰位点或标记物4. 引物序列的GC 含量一般为40-60%,过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大5. 引物所对应模板位置序列的Tm 值在72℃左右可使复性条件最佳。Tm 值的计算有多种方法,如按公式Tm=4(G+C)+2(A+T),在Oligo 软件中使用的是最邻近法(the nearest neighbor method)6. ∆G 值是指DNA 双链形成所需的自由能,该值反映了双链结构内部碱基对的相对稳定性。应当选用3’端∆G 值较低(绝对值不超过9),而5’端和中间∆G 值相对较高的引物。引物的3’端的∆G 值过高,容易在错配位点形成双链结构并引发DNA 聚合反应7. 引物二聚体及发夹结构的能值过高(超过4.5kcal/mol)易导致产生引物二聚体带,并且降低引物有效浓度而使PCR 反应不能正常进行 8. 对引物的修饰一般是在5’端增加酶切位点,应根据下一步实验中要插入PCR 产物的载体的相应序列而确定。 值得一提的是,各种模板的引物设计难度不一。有的模板本身条件比较困难,例如GC含量偏高或偏低,导致找不到各种指标都十分合适的引物;在用作克隆目的的PCR 因为产物序列相对固定,引物设计的选择自由度较低。在这种情况只能退而求其次,尽量去满足条件。 扩增较大片段DNA的PCR方法一般PCR方法在扩增大片段DNA时的局限性: 通常所用的PCR方法都在两个方面有局限,即目标产物精确程度和合成片段的大小。Pfu(Pyrococcus furiosus)DNA聚合酶,具有完整的3'外切酶校读活性(3'-editing-exonuclease),可以将每个循环中碱基的错配率由10*-4降到10*-3,从而提高PCR产物的准确性。但它在扩增1.5-2.0kb片段时,效率比Klentaq l(Taq DNA聚酶N-末端缺失突变体,类似于E.coli DNA聚合酶I Klenow片段)或AmpliTaq(全长的Taq DNA聚合酶)等聚合酶差;在扩增5.0-7.0kb片段时亦不比各种形式的Taq DNA聚合酶(如Ampli Taq、Klentaq 1、Klentaq 5等N-末端缺失的变异株)有明显优越之处。因而以往的PCR反应产物限制在5.0kb以内。超出这一范围,PCR扩增反应效率将明显下降,同时产物会降解。即使将延伸时间定为30分钟(10倍于通常所需)亦无改进。利用两种DNA聚合酶进行较大片段DNA的扩增 美国华盛顿大学医学院的Barnes WM等对前述问题进行了深入系统的研究,认为:PCR反应效率低最主要的原因是由于错配的碱基阻碍了延伸反应的正常进行,Pfu DNA聚合酶虽然可以通过“校读”功能纠正错配的碱基,但亦可能降解引物,尤其是在较长反应时间下;酶浓度较高时,反应效果更差。因此必需将Pfu DNA聚合酶的浓度控制在较低状态,同时配合使用Klentaq l等DNA聚合酶,这样既可以有效地去除错配,又可以使Klentaq l等催化的延伸反应顺畅进行。实验证实,按15:1 的比例混合使用Klentaq l和Pfu DNA聚合酶,引物大小为27-33nt,即可使反应有效进行。当然,对于各种不同条件的反应,两种类型酶的最佳配比需要具体考虑。控制脱嘌呤反应增强扩增效率 在PCR反应体系中某些成分耐热性较差,会影响反应效率。DNA聚合酶的热稳定性一般都是较好的,可能是模板DNA在温度较高的环境中某些位点发生脱嘌呤反应从而阻碍反应的顺利进行。Lindahl和Nyberg的研究结果显示:在70℃ pH7.4的条件下, 单链DNA脱嘌 呤反应的速度是双链DNA的4倍;100℃ pH7.0时,100kb的碱基中每分钟将有1个位点脱嘌呤。这一反应与缓冲体系中酸碱度的变化有关。人们注意到:三羟甲基氨基甲烷(Tris)的酸解离常数(pKa)会随温度升高而改变,平均每升高1℃,pKa值降低0.03。因而,在25℃时pH8.55的PCR反应体系,到95℃热变性时,pH值将变为6.45,这就很可能诱导脱嘌呤反应。 为了解决这一问题,可以采取下列措施: 缩短热变性时间,Barnes等在扩增35kb的大片段时,变性条件为95℃5秒,取得满意结果; 尽可能使升温、 降温过程缩短,可选择使用导热性能优越的薄壁反应管及较为先进的扩增设备; 适当提高反应体系的pH值,反应最初应控制在pH8.8-9.2范围; 适当增加延伸时间(可长至20分钟)。使用这种方法可以扩增最大为35kb的DNA片段,产物的准确性亦有充分保证,克服了以往基因克隆过程中出现的DNA分子内的碱基重排和可能的毒性危险等问题。 PCR常见问题PCR产物的电泳检测时间 一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。 假阴性,不出现扩增条带 PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量, ④PCR循环条件。寻找原因亦应针对上述环节进行分析研究。 模板:①模板中含有杂蛋白质,②模板中含有Taq酶抑制剂,③模板中蛋白质没有消 化除净,特别是染色体中的组蛋白,④在提取制备模板时丢失过多,或吸入酚。⑤模 板核酸变性不彻底。在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处 理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应 固定不宜随意更改。 酶失活:需更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而 导致假阴性。需注意的是有时忘加Taq酶或溴乙锭。 引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不 理想、容易弥散的常见原因。有些批号的引物合成质量有问题,两条引物一条浓度 高,一条浓度低,造成低效率的不对称扩增,对策为:①选定一个好的引物合成单 位。②引物的浓度不仅要看OD值,更要注重引物原液做琼脂糖凝胶电泳,一定要有 引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条 带,此时做PCR有可能失败,应和引物合成单位协商解决。如一条引物亮度高,一条 亮度低,在稀释引物时要平衡其浓度。③引物应高浓度小量分装保存,防止多次冻融 或长期放冰箱冷藏部分,







 我要推广仪器
我要推广仪器
 下载APP
下载APP