5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
需几个标准,哪位大侠有,帮偶找一下HG/T 3925-2007 甲基苯骈三氮唑 HG/T 2519-2007 工业六聚偏磷酸钠 HG/T 937-2007 工业用1,6己二胺 HG/T 3923-2007 循环冷却水用再生水水质标准 另还有四乙烯五胺的标准,多乙烯多胺非常感谢!
本人在做铁矿石全铁分析时,之前用的是氯化亚锡-三氯化钛方法测定全铁含量,用盐酸溶样180℃,10分钟左右,然后用氯化亚锡还原大部分三价铁,呈淡黄色后加钨酸钠,再用三氯化钛还原至乌兰。。。但是我发现在加钨酸钠的时候,会产生白色不溶沉淀,这种情况有时又不会产生,不知是什么原因,求指出问题!在加钨酸钠产生沉淀后我就用甲基橙作指示剂了,然后加氯化亚锡还原至甲基橙无色,冷却后,加硫磷混酸和二苯胺磺酸钠开始滴定,这样就省略了三氯化钛还原的步骤,不知用甲基橙作指示剂指示氯化亚锡还原三价铁终点可不可行,看到有文献这样做,而且比国标的方法简单,不知精确度怎么样,需要注意些什么?
三甲基氢醌与三甲基苯醌之间的关系,怎么计算两者之间的含量
我想问下大家,我用三甲基硅烷基重氮甲烷衍生青霉素,然后加入甲醇做催化剂,空白里没有青霉素,可是从峰上看,出现苯环结构,想问问有没有大神知道原因呢,求告知
[size=18px]目前在用AB的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测三苯基氯甲烷,Q1 MI模式扫243.1的离子[font=-apple-system, BlinkMacSystemFont, &](应该是三苯甲基碳正离子)[/font],发现基线非常高(30万-50万之间),且不稳定,时高时低,导致峰面积也 不稳定,打电话问客服,几个人几种说法,“液相部分污染了”“这个是正常现象,多走走就稳定了”,尝试用MRM模式去做,打出一个165.2的碎片,基线不到1000,做了线性和回收也都挺好,但是,这个碎片离子是怎么打出来的比较困惑,就怕以后再做的时候重现不出来……[/size][size=18px]流动相是90%甲醇,溶剂是正丁醇:乙腈(80:20)[/size][size=18px]请教一下各位大神,AB的仪器用SIM模式选择Q1 MI还是Q3 MI好呢?基线高且时高时低,除了污染还有什么原因呢?[font=-apple-system, BlinkMacSystemFont, &]三苯甲基碳正离子在质谱里能被打碎吗?会裂解成什么碎片离子?[/font][/size][size=18px][font=-apple-system, BlinkMacSystemFont, &][/font][/size]
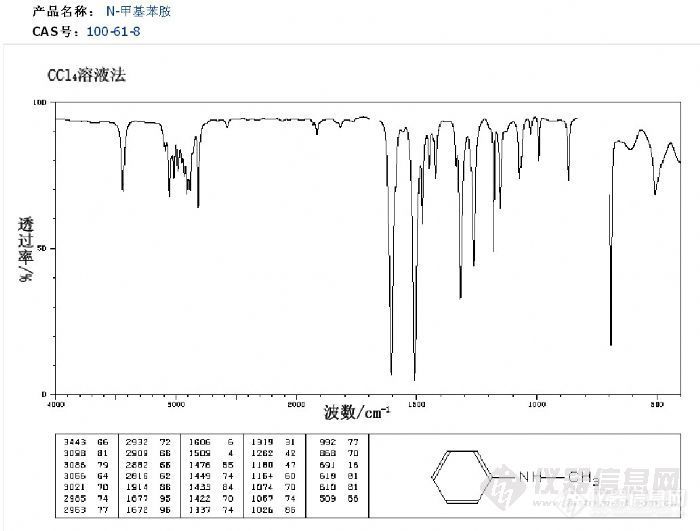
单位是做汽油检测的,最近要上新项目做汽油中N-甲基苯胺,甲缩醛2种添加剂,用的是中红外,初步查到的信息是N-甲基苯胺,的定性是在红外谱图中有检出甲基胺的吸收峰,则判断N-甲基苯胺检出。没接触过红外,不知道怎么看红外谱图中甲基胺的特征吸收峰。附上一张N-甲基苯胺的红外图。希望有懂的 给点建议http://ng1.17img.cn/bbsfiles/images/2012/03/201203221523_356642_2052186_3.jpg
液相色谱峰刚开始有正峰和反峰连在一起,乙腈和水做流动相的,是正常的吗,分析的2.4.6-三甲基苯甲酰氯含量甲醇酯化做的
关于有机硅分析微量水分,甲苯,甲基三乙氧基硅烷,氯化苯混合溶液怎么配[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]?
EP里面做N,N-二甲基苯胺的方法A里规定的毛细管色谱柱填料是:[color=#333333]交联聚甲基苯基硅氧烷(cross-linked polymethylphenylsiloxane R)。网上找了半天没找到具体型号,不知道大家有没有人知道这是什么型号的色谱柱?[/color]
我用滴定对苯醌的方法(溶液加碘化钾,盐酸,暗处静置后用硫代硫酸钠滴定)测三甲基苯醌含量,但是终点总是反色,找不到终点,请问高人们有解决的办法吗?谢谢!
柑桔中苯丁锡、三唑锡农药残留的检测 三唑锡和苯丁锡(FBTO)是两种常见的有机锡杀螨剂,可用于防治柑橘、苹果、棉花、蔬菜等作物的害螨,应用十分广泛。三唑锡和苯丁锡为难挥发化合物,不能直接用气相色谱法分析,需对其进行衍生化。文献报道苯丁锡的分析方法多为提取后用浓盐酸将其转化为氯化三苯基叔丁基锡,再用气相色谱-电子俘获检测器分析,但易受样品中共提取出来的电负性杂质的干扰。本文建立了柑桔中三唑锡和苯丁锡残留检测的气相色谱分析方法。柑桔中的三唑锡和苯丁锡经有机溶剂提取后,用甲基氯化镁衍生化为甲基三环己基锡(MeTCyT)和甲基三苯基叔丁基锡(MeTFBT),通过弗罗里硅土小柱净化后,用气相色谱-火焰光度(硫滤光片)进行分析测定。该方法操作简单,灵敏度高,检测结果准确可靠。1 材料方法1.1 仪器与试剂6890N气相色谱仪配火焰光度检测器(美国Agilent公司);CNM-MST-2多功能微量化样品处理仪(长沙中迅电子工程研制所);SK-1快速混匀器(金坛市高华仪器有限公司);TDZ4-WS低速离心机;正己烷(HPLC);丙酮(HPLC);乙酸(AR);乙醚(AR);0.5 mol/L硫酸(浓硫酸用水稀释);无水硫酸钠(AR);甲基氯化镁-乙醚溶液(22%,m/V,美国Aldrich公司);三唑锡(98%)和苯丁锡(96.5%)(德国Dr. S. Eherntorfer公司)。1.2 色谱条件色谱柱:HP-5 MS石英毛细管柱(30 m×0.32 mm(i.d)×0.25 μm);柱温:50℃保持1 min,以30℃/min 升至260℃,保持10 min;进样口温度:280℃;检测器(FPD,硫滤光片)温度:250℃;载气:N2,纯度≥99.999 %,1.5 mL/min;氢气流量:50 mL/min;空气流量:60 mL/min;进样体积:1μL;进样方式:不分流进样。分析步骤:[siz
我想了解4-氯-2-三氟甲基苯腈(4-Chloro-2-(trifluoromethyl)benzonitrile,CAS#320-41-2)的理化性质,但在网上只找到沸点109 º C (10 mmHg),是液体还是固体看不出来。因为这个沸点是真空条件下的。那位老师有相关的信息,请告诉我,谢谢!
300℃。含5mo1%苯基硅油的凝固点低达-70℃,表面张力约在2.1×10-4~2.85×10-4N/cm,相对密度1.00-1.11,折射率1.425~1.533。热稳定性好,250℃热空气中的凝胶化时间为1750h,还具有良好的耐辐照性能及高的氧化稳定性、耐热性、耐燃性、抗紫外性和耐化学性。可由八甲基环四硅氧烷、二甲基四苯基二硅氧烷、甲基苯基二乙氧基硅烷的水解物在催化剂存在下进行调聚反应来制取。用作润滑油、热交换液、绝缘油、气液相色谱的载体等。用于绝缘、润滑、阻尼、防震、防尘及高温热载体等,是电子仪表的理想液态阻尼介电液。http://www.zhongbaohg.cpooo.com/
求助给位,我想问一下四丁基溴化铵、十六烷基三甲基溴化铵、苄基三甲基溴化铵怎么做红外,它们都很容易潮解的,吸水性很强,我问一下,做红外的时候样品怎么处理,或者这些物质的标准红外数据在哪可以查到啊
请告诉2-甲基-4-甲氧基-二苯胺的液相色谱分析方法
谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法
有哪位大虾做过面粉中过氧化苯甲酰的检测?另,水产品中的甲基汞有做过检测的吗?如果是用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做的话,能不能把谱图发上来我参考一下,我急用。谢谢了!
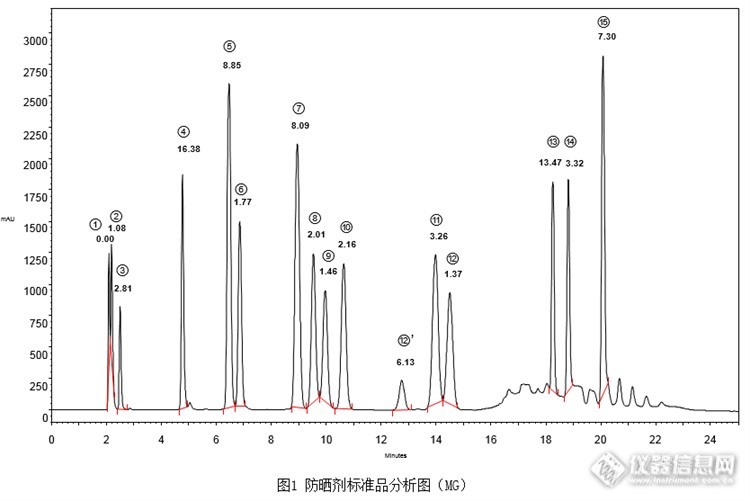
[align=center][b]2015版《化妆品安全技术规范》防晒剂检验方法-苯基苯并咪唑磺酸等15种组分[/b][/align][align=center][b]第一法(高效液相色谱-二极管阵列检测器法)[/b][/align]本次实验按照2015版《化妆品安全技术规范》中防晒剂检验方法的第一法(高效液相色谱-二极管阵列检测器法),对苯基苯并咪唑磺酸等15种防晒剂进行同时分析。15种防晒剂标准品按照《化妆品安全技术规范》配制成混合标准溶液,分别使用CAPCELL PAK C18 MG S5 4.6 mm i.d. × 250 mm,CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,CAPCELL PAK ADME S5 4.6 mm i.d. ×250 mm,CAPCELL PAK C18 AQ S5 4.6 mm i.d. × 250 mm以及SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm五款色谱柱对混合标准溶液进行分析。其中,MG和MGII色谱柱得到相对较好结果,但两款色谱柱原流动相条件下,个别峰未实现基线分离。结果如图1、图2。[img=,690,460]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_01_2222981_3.png[/img][img=,690,432]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_02_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,304]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_03_2222981_3.png[/img]为得到更好的分离效果,使用1支更新的MGII色谱柱,在原流动相条件基础上,对梯度进行调整,结果如图3所示。各峰分离度得到明显改善,但峰11和峰12分离度为1.43,仍未达到基线分离。[img=,690,425]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170933_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,292]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170933_02_2222981_3.png[/img]继续调整梯度条件,分析结果如4所示。在此条件下,各峰实现基线分离,得到良好分析结果。[img=,690,421]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170935_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,307]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170937_01_2222981_3.png[/img]接下来将色谱柱更换为MG色谱柱,在调整后的梯度条件下进行分析,结果如图5所示,同样可得到良好的分析结果。[img=,690,419]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170938_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,291]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170940_01_2222981_3.png[/img]
测定多环芳烃,用六甲基苯溶液作内标,购买的六甲基苯是固体的,请教溶液怎样配?是不是用二氯甲烷做溶剂?具体怎样操作,溶质和溶剂的量是多少?E-mail:zhoujuan@stu.xjtu.edu.cn
请教各位大侠有没有做:臭氧危害物質(CFC/HCFC/Halon),氟溴烃,單甲基二氯二苯基甲烷,單甲基二溴二苯基甲烷 (DBBT),單甲基四氯二苯基甲烷 的检测方法,有的话能不能给我传一份,我的mail是wei.gao@isti.ocm.cn。万分感谢!
我们做对氟苯甲酸检测,别人推荐用N,O-双(三甲基硅基)乙酰胺反应后测他们的衍生物,可是我搞不明白这个反应原理,不知道是不是可以反应彻底,请诸位高手帮忙讲解一下,谢谢!
群友问:用GB 5413.25-2010 第二法气相色谱法做肌醇,实验室买的三甲基氯硅烷和六甲基二硅胺烷有本底干扰,不知道老师们这两个试剂买的是什么牌子的?群友一:国药的群友二:三甲基氯硅烷,扫尾剂级别,100ml,国药有的,六甲基二硅胺烷,国扫尾剂级别,25ml,也是国药的;这两个试剂SIGMA有专用的衍生剂级别的。非常感谢乳制品检测之家群友岑老师、人气老师的分享!
2-甲基苯并咪唑和邻硝基苯胺的的紫外吸光度事多少?
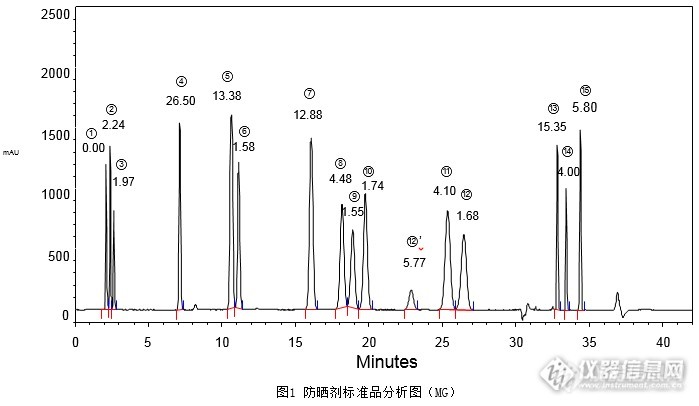
[align=center][b]2015版《化妆品安全技术规范》防晒剂检验方法[/b][/align][align=center][b]苯基苯并咪唑磺酸等15种组分-二元梯度法[/b][/align][align=center][b] [/b][/align]在2015版《化妆品安全技术规范》防晒剂检验方法中,第一法对15种防晒剂的分析为三元梯度方法,此法要求仪器配备三元泵,且四氢呋喃会对PEEK基材的管路和仪器有溶胀作用,所以该方法在实际操作上会受到一定的限制;而第二法将15种防晒剂分为两组,在不同流动相条件下分别检测,较为费时费力,且前12种防晒剂峰未能达到基线分离。基于以上情况,本次实验采用二元梯度方法,对15种防晒剂标准品进行同时分析,既可在常规二元泵系统进行实验,也可免去分组分析的繁琐。本实验混合标准溶液按照《化妆品安全技术规范》配制,分别使用资生堂CAPCELLPAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 250 mm和CAPCELL PAK C[sub]18[/sub] MGII S5 4.6 mm i.d.× 250 mm色谱柱进行分析,结果如图1和图2所示,两款色谱柱在二元梯度条件下均可使15种防晒剂峰实现基线分离。[img=,690,400]http://ng1.17img.cn/bbsfiles/images/2017/08/201708230913_01_2222981_3.png[/img][img=,690,366]http://ng1.17img.cn/bbsfiles/images/2017/08/201708230913_02_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,629,207]http://ng1.17img.cn/bbsfiles/images/2017/08/201708230913_03_2222981_3.png[/img]
RT?新购的气相色谱仪和柱子,只有上面两根柱子,一根是准备做中药材 醇类含量,一根是做农残的,省检定中心要来进行仪器检定,提前让我们准备好能做 正十六烷的 柱子,我想问下50%苯基甲基聚硅氧烷 能不能做?查阅资料,能做 烷烃的 有100%甲基聚硅氧烷(胶体和流体)、5%苯基聚硅氧烷, 我们现在有的PEG和 50%苯基甲基聚硅氧烷 能否做?万分感谢!
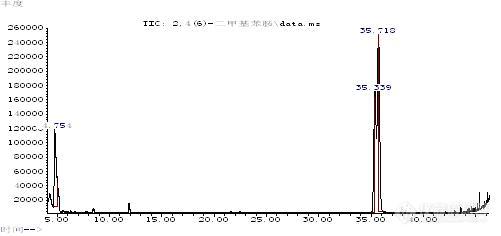
2,4-二甲基苯胺和2,6-二甲基苯胺的鉴别2,4-二甲基苯胺和2,6-二甲基苯胺同属于国家强制标准GB18401-2003附录C中所列的还原条件下染料中不允许分解出的23种芳香胺之一,二者又属于同分异构体,沸点和极性都很接近,故在检测过程中很难鉴别。目前,对于两者的分离鉴别主要靠液相色谱来实现,而使用气-质联用仪来鉴别两者还没有很好的方法。而针对有害芳香胺的气相色谱-质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。由于2,4-二甲基苯胺和2,6-二甲基苯胺沸点太接近,单纯依靠两者的沸点差异来实现其分离鉴别是有一定难度的。于是,作者考虑采用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),除了利用2,4-二甲基苯胺和2,6-二甲基苯胺的沸点差异外,再利用中等极性柱对于二者的保留作用差异来研究二者的分离鉴别。通过改善优化色谱条件,作者使用中等极性色谱柱DB-17MS,同时使用三阶程序升温,实现了2,4-二甲基苯胺和2,6-二甲基苯胺的较好分离。1 试验1.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)叔丁基甲醚 分析纯 国药集团化学试剂有限公司甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂2,4-二甲基苯胺和2,6-二甲基苯胺均为德国Dr.Ehrenstorfer公司。1.2 试样的制备分别称取适量的2,4-二甲基苯胺和2,6-二甲基苯胺,以甲醇为溶剂分别配制适宜浓度的2,4-二甲基苯胺溶液、2,6-二甲基苯胺溶液和2,4-二甲基苯胺和2,6-二甲基苯胺混合溶液。1.3 仪器操作条件色谱柱:DB-17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至[/font
各位老师 我想 求一下 甲基肼, 三氯化磷,磷化氢的标准曲线和相对应的吸光度 谢谢 甲基肼-对二甲胺基苯甲醛分光光度法 三氯化磷-对氨基二甲基苯胺分光光度法 磷化氢-钼酸铵分光光度法 五氧化二磷-钼酸铵分光光度法谢谢各位老师
小弟做了好几次2-溴,3,3‘-二甲氧基联苯的氢谱,都发现甲基有三重峰,做何解?谢了!
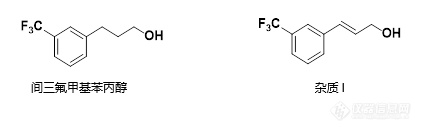
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]







 我要推广仪器
我要推广仪器
 下载APP
下载APP