乙酸对叔丁基环己酯 和乙酸邻叔丁基环己酯如何区分?谢谢
今天别人送个高效氯氰菊酯来给我分析我查行标是 用正己烷+无水乙醚=98+2 做流动相因为我没有无水乙醚 请问我可以用乙酸乙酯或者四氢呋喃代替吗代替后的 配比是多少啊 请高手解答!
请问冰乙酸和无水冰乙酸是一样的吗?
各位大侠:请问一下 GB/T 18446-2009 的 5.1 条乙酸乙酯无水,是指的多少水份呢?要是用分子筛除乙酸乙酯里面的水份的话,要加多少呢?谢谢!
近日在摸索无水乙酸钠含量测定,但是做了几天了,还没有做出来!无水乙酸钠含量测定需称取试样,置于铂皿中缓缓加热炭化,灼烧至白,冷却,溶于100ml热水中,加10滴溴甲酚绿-甲基红指示液,用盐酸标准滴定溶液(0.5mol/L)滴定至溶液由绿色变为暗红色,煮沸,冷却,继续滴定至溶液呈暗红色。但是操作中样品熔融以后就很难成固体,也就是无法灼烧至白,我试过放冷凝固后进行滴定,可是滴定过程中颜色不好判断,而且含量不合格,才百分之八十多。请各位大侠指教!
求助三氯吡氧乙酸、2,4-二氯苯氧乙酸正丁基酯(2,4-D丁酯)的检测方法
告急:哪位知道新版本测定乙酸正丁酯的国标标准。多谢!
我现急需乙酸丁酯的毒理性资料,不知有哪位大侠珍藏.能否奉献出来让我救救急啊?某某在此先道声:谢谢!!!AHCLRM-1@163.COM
用乙酸和丁醇反应,最后蒸馏的温度(124)上不去是啥原因急急!!
HGT 3498-1999 化学试剂 乙酸丁酯.pdf[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=39316]HGT 3498-1999 化学试剂 乙酸丁酯.pdf[/url][em07]
乙酸丁酯是不是用途特别广泛?
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
乙酸乙酯沸点77.06℃,折光率1.372 3,相对密度0.9003。乙酸乙酯一般含量为95%~98%, 含有少量水、乙醇和乙酸。可用下法纯化:于1000mL乙酸乙酯中加入100mL乙酸酐,10滴浓硫酸,加热回流4h,除去乙醇和水等杂质,然后进行蒸馏。馏液用20~30g无水碳酸钾振荡,再蒸馏。产物沸点为77℃,纯度可达以上99%。
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
各位大侠:本人最近的色谱出现故障,在分析室内空气tvoc,经过采样管分析样品时,乙酸丁酯几乎不出峰,但使用直接液体进样则不会出现这样的故障。我是使用热解析分析采样管的,解析结束后手工进样大家解析的条件是什么啊!急急急急急急
(1)原理试样经处理后,在pH6左右的溶液中,镉离子与二硫腙形成配合物,并经乙酸丁酯萃取分离,导入原子吸收仪中,原子化以后,吸收228.8nm共振线,其吸收值与镉含量成正比,与标准系列比较定量。(2)试剂 氨水、混合酸、1g/L二硫腙-乙酸丁酯溶液(称取0.1g二硫腙,加10mL三氯甲烷溶解后,再加乙酸丁酯稀释至100rnL,临用时配制)、2mo1/L柠檬酸钠缓冲液(称取226.3g柠檬酸钠及48.46g柠檬酸,加水溶解,必要时加温助溶,冷却后加水稀释至500mL,临用前用1g/L二硫腙-乙酸丁酯溶液处理以降低空白值)、镉标准储备溶液和标准使用液的配制与碘化钾-4-甲基戊酮-2法中的相同。(3)仪器原子吸收分光光度计。(4)分析步骤①试样处理对于谷类要去除其中的杂物及尘土,必要时除去外壳。对于豆类,取可食部分洗净晾干,切碎充分混匀。②样品消化称取5.00g上述试样,置于250mL高型烧杯中,加15mL混合酸,盖上表面皿,放置过夜,再于电热板或电砂浴上加热。消化过程中,注意勿使干涸,必要时可加少量硝酸,直至溶液澄明无色或微带黄色。冷后加25mL水煮沸,除去残余的硝酸至产生大量白烟为止,如此处理两次,放冷。以25mL水分数次将烧杯内容物洗入125mL分液漏斗中。取与处理样品相同量的混合酸、硝酸按同一操作方法做试剂空白试验。③萃取分离 吸取0、0.25mL、0.50mL、1.50mL、2.50mL、3.50mL、5.0mL镉标准使用液(相当于0、0.05μg、0.1μg、0.3μg、0.5μg、0.7μg、1.0μg镉)。分别置于125mL分液漏斗中,各加盐酸(1+11)至25mL。向试样品处理溶液、试剂空白液及镉标准溶液各分液漏斗中各加5mL柠檬酸钠缓冲液(2mol/L),以氨水调节pH至5~6.4,然后各加水至50mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),以氨水调节pH至5~6.4,然后各加水至501mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),振摇2min,静置分层,弃去下层水相,将有机层放入具塞试管中,备用。④测定测定方法与碘化钾-4-甲基戊酮-2法中的相同。⑤结果计算 样品中镉的含量按下式进行计算。X=/(m×1000)式中,X为试样中镉的含量,mg/kg;A1为测定用试样液中镉的质量,μg;A2为试剂空白液中镉的质量,μg;m为试样质量或体积,g或mL。计算结果保留两位有效数字。⑥精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
标准配法是吸取2ml乙酸正丁酯用60%乙醇定容到100ml,怎么从安瓿瓶中吸2ml出来?移液管都比安瓿瓶粗啊?
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
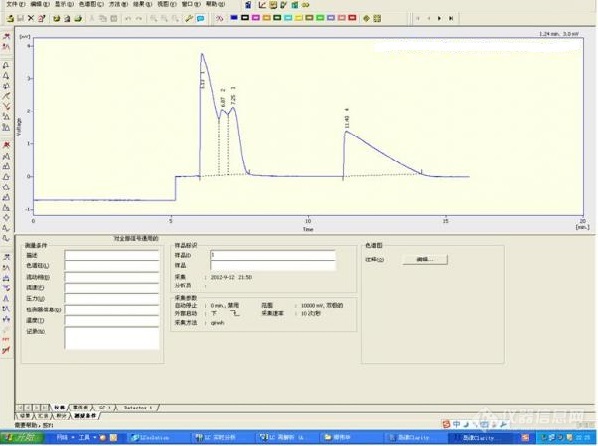
[color=#444444] 体系:[/color][color=#444444]乙酸/正丁醇/乙酸正丁酯/水;[/color][color=#444444]质量分数:均为25%[/color][color=#444444]沸点:乙酸119℃,正丁醇118℃,乙酸正丁酯126℃,水100℃。[/color][color=#444444] 分析条件:[/color][color=#444444]1. 色谱柱:DB-FFAP (30×0.25×0.25)[/color][color=#444444]2. 检测器:TCD[/color][color=#444444]载气流量:N2,0.25ml/min[/color][color=#444444]进样口温度:150℃[/color][color=#444444]柱温箱温度:120℃[/color][color=#444444]检测器温度:220℃[/color][color=#444444]图上可以看出:[/color][color=#444444]1, 基线发生瞬间跳动,一般基本上是漂移吧,为什么我这会突然发生阶跃式的跳动?分析原因是不是载气流量太低,可能载气流量发生不稳定,导致基线发生跳动?[/color][color=#444444]2, 由于有四元组分,前三个峰没有完全分开,而色谱柱分离效率跟载气流量,柱温关系最大,载气流量已经很低了,而且柱温也低于乙酸正丁酯的沸点,是不是也不能再低了。考虑能不能采用程序升温的方法,有没有可能将峰分开?如果可以的话,程序升温建议应该如何设置?[/color][color=#444444]求高手。[/color][color=#444444][img=,598,446]https://ng1.17img.cn/bbsfiles/images/2019/09/201909271014211801_6141_1752329_3.jpg!w598x446.jpg[/img][/color]
有高手做过乙酸丁酯没有,给个方法,最好能附上图谱
[size=4]GB-T 603-2002 化学试剂 试验方法中所用制剂及制品的制备 里面的乙酸---乙酸钠缓冲溶液配置是用三水乙酸钠,可是我手头没有三水的,只有无水乙酸钠。我按重量换算成无水乙酸钠的重量配置出的缓冲溶液可不可以呢?[/size]
求助各位了,现在我需要用液相色谱定量分析氯乙酸和氯乙酸丁氧基乙酯的混合体系中氯乙酸的含量,选择什么紫外波长较为合适呢?200nm或者210nm可以吗?
我一向认为乙酸乙酯卡尔费休滴定用普通试剂就可以了,有个羰基不等于醛酮.RdH的应用手册上也说直接滴定就可以了。但是梅特勒和万通的人都说要用醛酮试剂,有道理吗?
各位专家,大家好,我现在用GC112A分析乙酸丁酯和2,3-丁二醇,柱子用的是SE-30和SE-54,2,3-丁二醇沸点为181左右,乙酸丁酯为126.1,可是乙酸丁酯的峰在2,3-丁二醇后面,而且分离度不够,不能大于1.5,我采用了各种升温程序都不行,有谁能告诉我怎么办么??
我们生产工业乙酸乙酯,乙醇和乙酸酯化得到的。中控分析用的是TCD(因为要分析出水分含量)。做校正因子的时候遇到以下情况第一种方法:买的试剂(无水乙醇,乙酸乙酯分析纯)因为含有水分所以我用分子筛吸附了水分,吸附好以后在库仑水分仪上测得水分0.001%。然后在天平上配置标液。做校正因子,用校正面积归一法定量第二种方法:买的试剂不经分子筛去水直接配置标液做校正因子,用校正面积归一法定量上述两种定量方法测得的乙醇相对乙酸乙酯的校正因子差了很多。第一种测得是f=1.6,第二种0.7,查文献是0.8..想不通什么原因,难道是分子筛处理的原因。请专家指教。谢谢
[color=#444444]我想用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分离乙酸丁酯-水的混合液,乙酸丁酯的浓度低于1%,0.1%-1%的含量。已经把分流比调到60:1,流速小于1,温度:30,60,70,100,都试过了,可是低浓度的还是分不开,想麻烦问一下有木有人测过。谢谢啊![/color]
各位同仁: 我使用的是北分sp3420气相色谱,分析条件是:90度保留8.5分钟,每分钟50度升至终温250度,保留2分钟。我进tvoc标样时乙酸丁酯不出峰是怎么回事啊
最多用30ML乙酸丁酯萃取过2.5g镓,但不知道最大量是多少?有人知道乙酸丁酯能萃取镓的最大量吗?谢谢
我公司做循环冷却水中的铜离子测定?根据国家标准《GBT 13689-2007 铜的测定》要用四氯化碳进行萃取!!!可四氯化碳毒性太大了,打算尝试用乙酸丁酯代替之。在这里乙酸丁酯能代替四氯化碳作萃取剂嘛?望高手解答。如有关于有机萃取剂的资料 也希望能共享下。
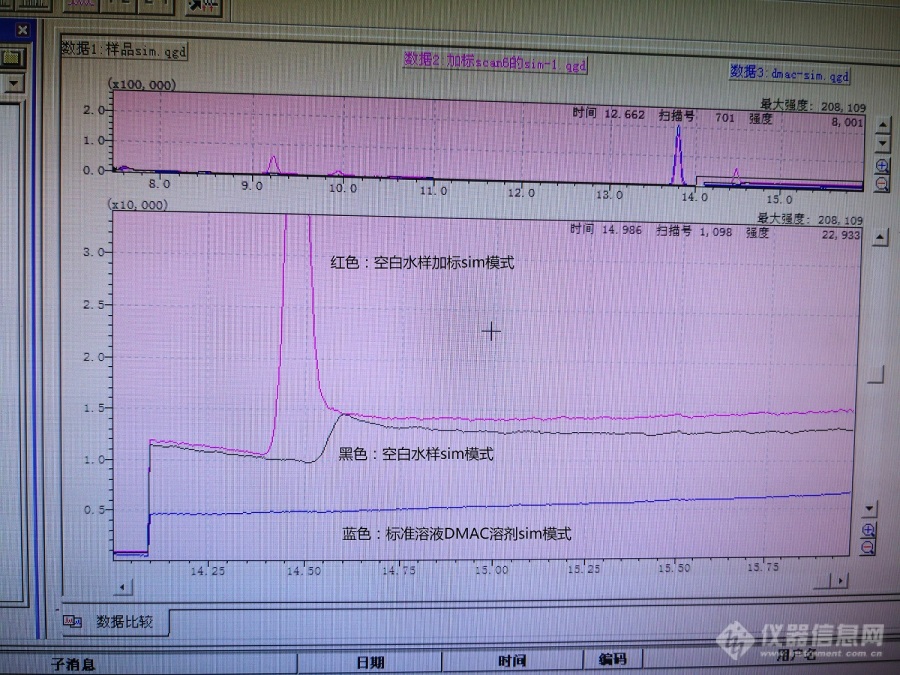
[font=宋体]用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]新建水样中乙酸丁酯等检测方法,柱子[/font]wax[font=宋体],程序升温[/font]60[font=宋体]℃([/font]8.5min[font=宋体])[/font]-5[font='微软雅黑','sans-serif']℃[/font][font='微软雅黑','sans-serif']/min-70℃-10℃/min-200℃-20℃/min-250℃[/font][font=宋体]([/font][font='微软雅黑','sans-serif']5min[/font][font=宋体]),乙酸丁酯出峰时间[/font][font='微软雅黑','sans-serif']14.6min[/font][font=宋体]。用母液(溶剂[/font][font='微软雅黑','sans-serif']DMAC[/font][font=宋体])进样峰型都很好。但是进样线性溶液(取母液用水稀释)、加标样品(母液加水样)以及溶剂水,基线就会在乙酸丁酯位置出现突越(见下图[/font][font='微软雅黑','sans-serif']14.6min[/font][font=宋体])。调整升温速率,突越总是跟乙酸丁酯一起。也老化过柱子,没有改善。请教各位老师,该突越是基线问题还是有杂质干扰,该如何去除。[/font][font=宋体][img=,656,491]https://ng1.17img.cn/bbsfiles/images/2020/03/202003261051583378_4789_2332103_3.jpg!w690x517.jpg[/img][/font]







 我要推广仪器
我要推广仪器
 下载APP
下载APP