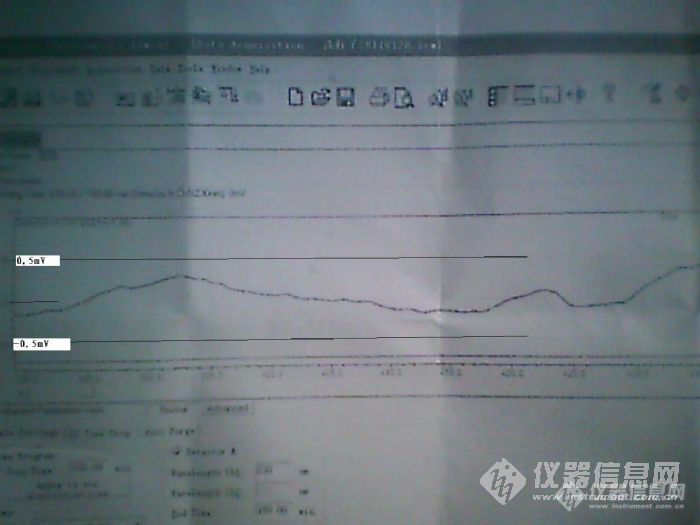
向各位高手请教事情的经过时这样的2011年1月18日,岛津液相,流动相是甲醇:20m mol/L 磷酸二氢钾溶液(22:78),在跑样的过程中突然断电,(没有关仪器),几分钟后突然来电又断电,(此时,关闭仪器)。于是去找工程部,工程部说只能供应30分钟,于是抓紧时间用5%甲醇洗脱30分钟(此时没有开空调,温度可能只有几度,跑样过程时候温度为25度).1月19日,开机首先用5%甲醇系统三个小时,再用水醇,甲醇洗脱,基线漂移厉害,那时候想可能柱子坏了,于是关机。此后几天没有人用此台仪器。1月26日,用水醇,甲醇洗脱一天,基线朝一个方向漂移厉害,始终不平(这时想可能检测器污染了)。1月27日,不接柱子,用纯水洗脱三个小时,然后过渡到甲醇,再用甲醇洗脱,基线与1月26号比较明显改善,但仍然不稳定,30分钟基线漂移1.5mV左右。1月28日,不接柱子,首先用纯水洗脱,再用甲醇洗脱,再用水洗脱,再用异丙醇洗脱,再用甲醇洗脱,基线仍然不理想。截下一段50分钟的基线,如图请问是仪器本省硬件元件的问题还是检测池污染啊?十分感谢!http://ng1.17img.cn/bbsfiles/images/2017/01/201701191652_630081_2222645_3.jpg
请问: 我以前用FFAP柱子做甲醇,条件已经调好了,进甲醇标液峰形都很好,但最近再进甲醇标液,无论进多大浓度,峰都拖尾的很严重,请问是怎么回事?应该如何改善?
甲醇介质的呋喃丹标准曲线系列能保存使用多长时间?望老师不吝赐教
请问甲醇的截止波长是多少?
做酒剂里面的甲醇量测定,甲醇峰峰型每针都变化,不知道问题出在哪,一会儿拖尾大一会儿拖尾小,柱温程序升温(见图片2),进样口分流比30,温度200,FID220,BD624的毛细管柱[img]https://ng1.17img.cn/bbsfiles/images/2022/04/202204061337444493_9830_3475778_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/04/202204061337444205_5246_3475778_3.png[/img]
我看培训资料说进质谱摸离子条件最好用50%甲醇水或50%乙腈水,文献里也有报道50%甲醇水溶解配制储备液的,但是我自己做的时候发现50%甲醇水里药品不溶,超声了10分钟之后也是。那我这个储备液算是废了吗,是不是得重新配储备液了?还能补救吗?我做的是联合检测,多个药品的溶解溶剂不同会不会显得我这个方法不够简便呢?如果全部改用甲醇溶解和稀释会不会不好?纯甲醇进质谱摸条件说是不容易电离。
我们实验室有三台分析甲醇的气相色谱,用的色普柱都是wax柱,进样方式有阀进样(气体中甲醇分析)和微量针进样(水中甲醇分析),但都有一个共同的特点就是峰拖尾,如果把正常峰形比作等腰三角形的话,那么甲醇峰形就是直角三角形。想问一下用过wax柱的有没有 同样的问题。
汽油中甲醇峰拖尾不尖锐,其它峰正常,怎么办?
各位大侠,小妹在做酒中甲醇时,甲醇峰有点拖尾,我用的是3米的填充柱,进样量是0.5微升。
安捷伦7890B,阀进样。分析甲醇时峰出[img=,546,415]https://ng1.17img.cn/bbsfiles/images/2018/12/201812050949395687_7589_3323561_3.png!w546x415.jpg[/img]现梯形拖尾,几ppm至几千ppm的氮中和空气中甲醇都有严重拖尾。
请教一下 固相萃取新烟碱类农药的文献中大部分用的甲醇 但是也有不少用的氨水甲醇 洗脱效果也比较好…请问用氨水甲醇的原理是啥?为啥要加氨水在洗脱剂里
7890A气相刚装没多久,现在用DB-624做一个样品的溶剂残留,直接进样的,测得是甲醇和乙腈,溶剂用的是DMF,配好后进样为什么甲醇和乙腈都会拖尾?看到网上的经验甲醇和乙腈的分离应该挺好的呀!对了,80C,1.5ml/min条件下5 6 分钟出峰,两个峰份的挺好,就是拖尾!求助大神们能解决一下小弟的麻烦!不胜感激!
请问用甲苯溶解的样品可以进液相用甲醇或乙睛的流动相洗脱吗?

按药典四部通则中辅料乙醇检查挥发性杂质,方法如下(药典标准),结果就甲醇峰拖尾(4.12那个峰为甲醇峰),其余峰的峰型都很好,柱子换新的还是这样,求助,是甲醇不适合在中极性柱子上分离才导致峰拖尾的吗?大家有遇到过吗?怎么解决的?[img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905070838480838_5186_3480890_3.jpg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905070840080160_3142_3480890_3.jpg[/img]
7890测白酒中甲醇含量响应低且拖尾 条件如下:毛细管柱:CP-Wax 57 CB acidic 50m×0.25mm×0.2μm进样口温度: 200℃ 分流比:30:1检测器温度: 220℃进样量:1μl程序升温:40℃保持5分钟,10℃/min 升至180℃保持5分钟如何解决响应低且拖尾的问题?
仪器条件都没有差别,进样也是连续的纯的样品出峰形好 对称用甲醇稀释后进样的峰形就前拖尾很严重,甲醇峰形也是好的对称的。样品和甲醇不会反应 也没有共沸现象。怎么回事??
我最近实验发现甲醇乙腈混合的话(不含水),不论什么比例貌似都比纯乙腈洗脱能力强。即洗脱能力:纯甲醇纯乙腈甲醇乙腈混合溶剂 不知大伙对乙醇丙酮的洗脱力有没有研究 还有通常情况下乙腈分离度比甲醇要好(甲醇分不开的情况乙腈可以分开若对此有不同观点我也不反对毕竟甲醇也有其特殊性),我想知道有没有比甲醇乙腈让样品间得到更好分离度的流动相啊?
各位大神,帮帮忙吧,做苯系物的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测,标品是溶在甲醇里的,但甲醇峰拖尾太严重,把苯峰覆盖了,想用二硫化碳来稀释标品,但是两者不互溶呀?怎么办呀怎么办?
看到论坛上关于乙腈和甲醇的洗脱能力的疑问,特此发表下甲醇/水 乙腈/水 四氢呋喃/水10 相当于 6 相当于 420 12 1030 22 1740 32 2350 40 3060 50 3770 60 4580 73 5390 86 63100 100 72溶剂混溶性表溶 剂 极性指数 折光度20℃ 紫外截止波长 沸 点(℃) 黏 度(cPoise) 水中溶解度(%W/W) 乙酸 6.2 1.372 230 118 1.26 100 丙酮 5.1 1.359 330 56 0.32 100 乙腈 5.8 1.344 190 82 0.37 100 苯 2.7 1.501 280 80 0.65 0.18 正丁醇 4.0 1.394 254 125 0.73 0.43 四氯化碳 1.6 1.399 263 77 0.97 0.08 氯仿 4.1 1.466 245 61 0.57 0.815 [color=black
分析废气中的苯系物时,我们用的国标的萃取溶剂是二硫化碳(活性炭吸附 二硫化碳解析)。分离效果很好。现在需要使用甲醇作溶剂,甲醇极性很强,作溶剂时拖尾问题严重。一直无法解决。我试过很过毛细管色谱柱,极性非极性都试过了。如HP-5,HP-FFAP,PEG20M,HP-1701 都无法解决甲醇的拖尾问题。PS:仪器各条件设定。各路气体流速,流量都没问题。请高手指点!万分感谢~
背景:1、物质必须要用气质进行分析,我用的HP-5柱2、物质极性较大,液液萃取方法不适用3、只能采用SPE的方法,甲醇为洗脱溶剂4、最后如果对甲醇做换相处理,目标物损失较大 最近遇到这个问题,所提取的物质只能用甲醇作为溶剂进气质,论坛及各方面都查过,对于甲醇是否能进气相的说法不一致,有说甲醇会损伤柱子不能做进样溶剂的,也有人说可以,因此再把这个问题提出来请教一下高手。 有没有人用甲醇做溶剂进过样本呢?是不是会把柱子搞废了。 谢谢!还请专家及有经验的老师给出解答!
求助:测定红色酒精饮料介质中的甲醇含量,可以不蒸馏用气相色谱直接进样测定吗?
DB624柱子,40度,4分钟运行。5:1分流,进样口250度,FID300度,载气为氮气,样品为10ppm、15ppm、20ppm的甲醇水溶液,进样体积1ul。拖尾老严重了,请问老师们,我该怎么办啊?先行谢过了!
[color=#444444]我最近在做阿托,发现配好的阿托放在冰箱里也会降解,溶剂用的是甲醇,看进口标准上用的是0.05mol/l的三羟甲基氨基甲烷:乙腈 1:1作为稀释液,有人考察过么 这个溶液会使阿托稳定吗[/color][color=#444444]还有弱弱的问一句正相色谱室不能走梯度的吧?[/color]
15%甲醇水溶液超声脱气会怎样

请教各位大侠一个问题:目前我在用NH2柱萃取乙烯利,具体程序如下: 活化:5mL甲醇 10mL0.5N的乙酸 平衡:10mL0.05N的乙酸 上样:2mL5ug/mL的乙烯利标准品 淋洗:10mL甲醇 洗脱:15mL1%TFA/甲醇经以上程序过柱子,并且分别对上样、淋洗、洗脱这三步的流出液进行收集,经40度旋蒸近干,用乙腈定容至1mL。GC分析,结果发现这三步都没有检出乙烯利,也就是说乙烯利都残存在柱子上面没有下来。但是根据乙烯利属强酸性物质的性质,选用NH2柱应该没有问题,百思不得其解,请固相萃取的高手指教吧!谢啦[img]http://ng1.17img.cn/bbsfiles/images/2010/04/201004270002_214892_1919829_3.jpg[/img]
15%甲醇水溶液超声脱气会怎样
大家用的色谱甲醇臭不臭啊?为什么我用的很臭呢?用的是天津的科密欧。本来是一直以为就是那样子,突然有一天一个学长说,越臭的越不好,他用的是进口的,就基本上没有味道。借此跟大家讨论一下,你用的色谱甲醇都是什么牌子的?臭不臭啊?:-)
为什么高效液相色谱要用甲醇和水做洗脱剂?
流动相是70%甲醇和30%水,进纯甲醇和水在同一时间出峰,但是流动相换成是70%乙腈和30%水,进纯甲醇时就会出来两个峰,这是怎么回事呢?麻烦大家能帮帮我呀,如果是溶剂峰的话为什么用乙腈是两个峰呢?我是色谱新手,多谢大家了







 我要推广仪器
我要推广仪器
 下载APP
下载APP