[size=5]超临界流体色谱法测定三七及云南白药中人参二醇和人参三醇的含量[/size] 来源: 作者:李云华,李修禄,虹岚,刘锦耀,张美义提要:本文用超临界流体色谱法(SFC用定了三七和云南白药中人参二醇及人参三醇的含量,使用15%硫酸乙醇-水(1 :1)直接进行水解,然后碱水解,克服了文献报道的水解不完全、结果不稳定.不能定量回收的缺点。本法取样量少,灵敏度高,操作简单,快速.整个分析过程可在8h内完成关键词:超临界流体色谱:人参二醇:人参三醇:三七:云南白药三七(Radix notogimeng)为我国名贵中药,其主要有效成分为人参皂甙,经强酸水解得到人参二醇(panaxadio1)和人参三醇(panaxatrio1)。三七及其中成药的质量控制一直是个难题,目前测定人参二醇及人参三醇的方法还存在不少问题:薄层层析的分离度不够理想并有杂质干扰:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法需要衍生化,操作复杂:高效液相色谱也需要衍生化。我们采用将样品直接强酸水解后用环己烷提取,经净化后,用超临界流体色谱法测定了三七及其中成药中人参二醇及人参三醇的含量,获得了较好的结果。实验部分一. 仪器和药品622型SFC—[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] 色谱仪(美国理氏科学仪器公司):HP 3390A 色谱数据处理机:SB-Cyanopropyl-50石英毛细管交联柱10m×50μm,膜厚0.25μm。人参二醇和人参三醇对照品购自中国科学院昆明植物研究所:三七粉、云南白药由昆明43医院药局提供。胆固醇为Merck公司产品,无水乙醇、二硫化碳、氢氧化钠,硫酸,环己烷均为分析纯试剂。二.色谱条件流动相为CO2,纯度99.995% ;FID 检测器,检测器温度300℃ ;柱温90℃ ;分流进样,进样量0.2μl分流比为l/1O:起始压力为lO.1MPa,程升1.01MPa/min至35.5MPa。记录仪参数:ZER0=0, ATT=2,CHT SP=0.4,THRSH=4,PK WD=0.2。
[color=#444444]HPLC测定人参皂苷Ra1对照品含量以色谱纯乙腈-水为流动相,为什么所得结果只有前五分钟的溶剂峰,却没有想要的保留时间为16分钟左右的峰呢?[/color]
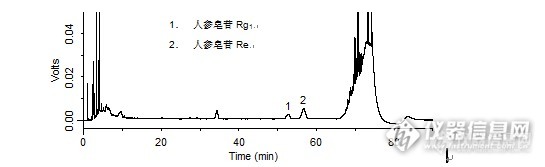
问题:启脾口服液中人参皂苷Rg1、人参皂苷Re的检测:对照品中人参皂苷Rg1、人参皂苷Re的分离度是多少?答案:2.467获奖名单:吕梁山(ID:shih20j07)dahua1981(ID:dahua1981)m3071659(ID:m3071659)http://ng1.17img.cn/bbsfiles/images/2016/02/201602231542_584931_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602231543_584932_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602231543_584933_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。启脾口服液中人参皂苷Rg1、人参皂苷Re的检测样品制备 制备方法1. 对照品:取人参皂苷Rg1对照品、人参皂苷Re对照品适量,精密称定,加甲醇制成每1 mL各含0.25 mg的混合溶液,摇匀,即得。2. 供试品:精密量取本品50 mL,加三氯甲烷振摇提取3次,每次30 mL,弃去三氯甲烷提取液,水液加水饱和正丁醇振摇提取5次(50 mL、30 mL、30 mL、20 mL、20 mL),合并正丁醇提取液,加氨试液洗涤4次,每次50 mL,弃去氨试液,再加正丁醇饱和的水轻轻振摇洗涤2次,每次50 mL,弃去水洗液,正丁醇液回收溶剂至干,残渣加甲醇溶解并转移至5 mL量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Diamonsil C18(2) 250 × 4.6 mm,5 μm (Cat#:99603) 流动相A:水 B:乙腈 梯度流速1.0 mL/min 柱温35 ℃ 检测器UV 203 nm 进样量5 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/02/201602231201_584890_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 52.603 387347 6585 18460.678 1.019 -- 2 56.668 262702 4169 16820.782 0.971 2.467 *药典要求理论板数按人参皂苷Re峰计算应不低于2500 供试品http://ng1.17img.cn/bbsfiles/images/2016/02/201602231202_584891_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 52.664 117855 2108 18875.714 0.982 -- 2 56.654 291453 4502 18266.289 1.019 2.486 *药典要求理论板数按人参皂苷Re峰计算应不低于2500本品种同时使用了Leapsil C18色谱柱,在药典规定条件下进行人参皂苷Rg1[/sub
现在我们公司增加辅料乙醇和醋酸钠的红外检测,需要购买红外乙醇对照品和醋酸钠对照品。请各位大侠提供除了中检所外,能够在一个星期内买到的厂家。谢谢大家了!!!!
中检所提供的乙醇对照品乙醇浓度是多少?
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。
问下:吸取的对照品无水甲醇、乙醛、乙缩醛是专门的对照品吗?还有用到的50μl、100μl、150μl取样管是微量进样器吗?
有没有人参加浙江省乳制品中三聚氰胺检测能力验证
我不小心那错了试剂,对照品用甲醇溶解,我拿了分析纯溶解,是否会影响结果?
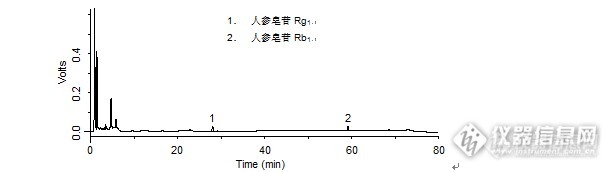
问题:心舒胶囊中人参皂苷Rb1、人参皂苷Rg1的检测:用到了迪马哪几款色谱柱?答案:Leapsil C18、Diamonsil C18、Platisil ODS获奖名单:捌道巴拉巴巴巴(ID:v3082413)大川之子,纵横四海(ID:chuangu120)zgx3025(ID:v2844608)http://ng1.17img.cn/bbsfiles/images/2016/03/201603211517_587699_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603211517_587700_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。心舒胶囊中人参皂苷Rb1、人参皂苷Rg1的检测样品制备 制备方法1. 对照品:取人参皂苷Rb1对照品、人参皂苷Rg1对照品适量,精密称定,加甲醇制成每1 mL各含0.2 mg的溶液,即得。2. 供试品:取本品20粒,精密称定内容物的重量,混匀,取约2 g,加乙醚约90 mL,加热回流1小时,滤过,滤渣及滤纸挥尽乙醚,置具塞锥形瓶中,精密加入甲醇25 mL,称定重量,超声处理(功率250 W,频率20 kHz)30分钟,放冷,再称定重量,用甲醇补足减的失重量,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Leapsil C18 100 x 4.6 mm,2.7 μm (Cat#:86002)流动相A:乙腈 B:水 梯度流速1 mL/min柱温30 ℃检测器UV 203 nm 进样量5 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603211119_587585_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 27.983 201139 11394 57017.208 1.004 -- 2 59.090 209117 15622 432268.026 0.951 75.113 *药典要求理论板数按人参皂苷Rb1峰计算应不低于5000供试品http://ng1.17img.cn/bbsfiles/images/2016/03/201603211119_587586_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 28.035 502352 27978 55489.037 1.042 -- 2 59.119 281333 20811 423485.934 0.946 74.059 *药典要求理论板数按人参皂苷Rb1峰计算应不低于5000本品种同时使用了Diamonsil C18、Platisil ODS两款色谱柱,在药典规定条件下进行人参皂苷Rb1、人参皂苷Rg1的检测,均满足药典要求。
有一个产品客户要求含量检项,没有对照品,需要用自己做的纯品来标化出对照品。根据合成人员提供的信息,用的乙醇作为溶剂,没有无机盐,含有水分。我标化时检测了纯度、水分,乙醇的残留,炽灼残渣也测了。最终得出一个含量,为99.4%。于是我拿这个含量试着测了一下样品,发现样品只有91%的含量……我又拿买的纯品,按照上面给的数值作为含量,测了一下样品,样品的含量达到了100%以上……很是蒙圈了- -今天把标化的对照品、买的纯品、样品,分别称取了10.00mg的样品,稀释至250ml,进样10ul,对比了一下三者峰面积。如果按照称样量与峰面积的比值来看的话,买来的纯品峰面积最高,是不是说买来的纯品含量应该也是最高的- -造成这种差异的原因是什么,能怎么处理呢?理论上要两种方式标化,但是这个化合物是吲唑上面挂了一个甲基和溴。还有其他方式可以标化吗?
药典 金钱草含量测定以甲醇一O.4%磷酸溶液(50:50)为流动相;检测波长为360nm,温度30℃。 对照品溶液的制备 取槲皮素对照品、山柰素对照品适量,精密称定,加80%甲醇制成每1ml各含槲皮素4μg、山柰素20μg的溶液,即得 供试品溶液的制备 取本品粉末(过三号筛)约1.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,密塞,称定重量,加热回流1小时,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过。精密量取续滤液25ml,精密加入盐酸5ml,置90℃水浴中加热水解1小时,取出,迅速冷却,转移至50ml量瓶中,用80%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。 本品按干燥品计算,含槲皮素和山柰素的总量不得少于O.10%。
请教各位:在乙醇的气相色谱检验中,对照品溶液制备用到的试剂:甲醇、乙醛、乙缩醛、苯、4-甲基-2-戊醇。这些试剂必须用国家标准品吗?还是能用色谱纯的试剂代替,如果能代替使用,需不需要做相关对比试验,又该怎么进行这些对比试验?谢谢各位大神解答。。。。
给位大神求帮我分析一下顶空进样测苯,苯对照品的溶剂是90%乙醇,加了10mg 的无水碳酸钠,三针对照总会有一针峰面积较小(只有一半),是什么原因?
请问老师,17有机氯农药混合溶液的对照品,是根据对照品说明书上出峰顺序定位的?各位老师,有谁做过人参农残,想问问大家是如何定位?
求金刚烷胺对照品 批号、纯度、厂商
其实我想这么做的原因就是想节约点成本,因为做西青果药材的,对照品没食子酸用50%甲醇溶解,样品也是用50%甲醇溶解,地榆这个药材也是没食子酸,但是浓度稍微低点,我就想用做西青果的对照稀释一下就好,免得再次称对照,地榆药材是用水处理的,药典上写着是称没食子酸适量,用水溶解,现在里面有甲醇,会影响准确性吗?
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo

问题:颈痛颗粒中三七皂苷、人参皂苷的检测使用了哪几款液相色谱柱?答案:Platisil ODS、Leapsil C18、Spursil C18-EP【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币mengzhaocheng(ID:mengzhaocheng)999youran(注册ID:999youran)WUYUWUQIU(注册ID:wulin321)http://ng1.17img.cn/bbsfiles/images/2016/01/201601291556_583923_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601291556_583924_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================颈痛颗粒中三七皂苷、人参皂苷的检测样品制备制备方法1. 对照品:取人参皂苷Rg1对照品、人参皂苷Rb1对照品和三七皂苷R1对照品适量,精密称定,加甲醇制成每 1 mL含人参皂苷Rg1 0.5 mg、人参皂苷Rb1 0.5 mg、三七皂苷R1 0.1 mg的混合溶液,即得。2. 供试品:取装量差异项下的本品适量,研细,取约1.3 g,精密称定,加乙醚40 mL,加热回流30分钟,滤过,弃去乙醚液,药渣及滤纸挥尽乙醚,再精密加入甲醇40 mL,称定重量,加热回流1小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液20 mL,回收溶剂至干,残渣加水10 mL使溶解,用水饱和的正丁醇振摇提取5次,每次10 mL,合并正丁醇提取液,用2%碳酸钠溶液洗涤2次,每次20 mL,再用正丁醇饱和的水洗涤2次,每次20 mL,取正丁醇液回收溶剂至干,残渣用甲醇溶解,转移至10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得。分析条件色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相A:水 B:乙腈 梯度流速1.0 mL/min柱温30 ℃检测器UV 203 nm进样量10 μL色谱图对照品 http://ng1.17img.cn/bbsfiles/images/2016/01/201601290942_583886_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 36.408 264983 25847 265850.102 1.018 -- 2 38.915 1775914 178004 318998.439 0.998 8.983 3 54.506 1316933 127848 597818.242 0.870 55.926 *药典要求理论板数按三七皂苷 R1峰计算应不低于3000供试品http://ng1.17img.cn/bbsfiles/images/2016/01/201601290943_583887_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 36.344 250230 25147 274216.739 0.996 --
请问老师配苍术素对照品时,用甲醇为什么溶解不了,一直有悬浮的颗粒
请问配苍术素对照品时,用甲醇为什么溶解不了,一直有悬浮的颗粒
各位老师:乙醇用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测挥发性物质时,进对照品a出来的峰拖尾还分不开是什么原因呢[img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012387514_2300_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012529407_1811_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012539217_9202_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012593807_5993_3926493_3.jpeg[/img]
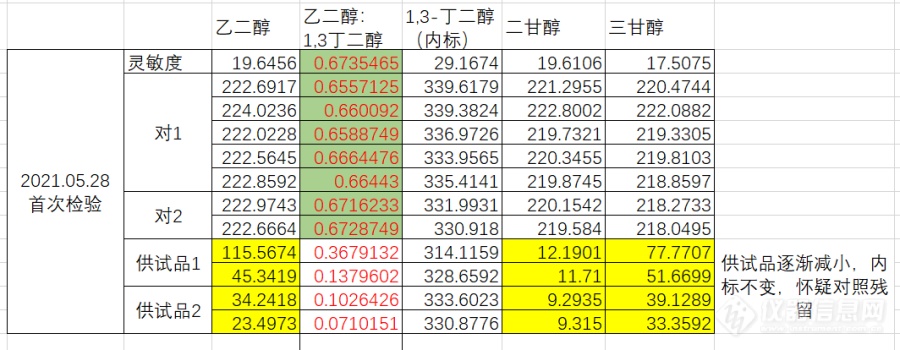
在进行聚山梨酯80Ⅱ 乙二醇、二甘醇和三甘醇检测过程中,发现对照品溶液重复性相当不错,但是供试品中目标峰逐渐减小,见如下数据:[img=第一次实验时实验数据,690,268]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031131180784_3076_3356999_3.png!w690x268.jpg[/img]当时出现这种情况,便怀疑是对照品残留到了供试品中,然后减少了进样针数,实验数据如下:[img=减少对照进样次数,690,147]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031133386103_879_3356999_3.png!w690x147.jpg[/img]供试品确实小了很多,而且也没有逐渐减小的情况,当时基本确定是对照品残留,因此后续实验调整了进样顺序,首先进供试品,但是又出现跟首次检验一样的情况,供试品目标峰面积逐渐减小,而后面的对照重复性也是相当不错做到此时怀疑是色谱柱问题,因此新采购了色谱柱,但供试品逐渐减小的问题仍然存在,后续又按照空白2针,供试品2针,空白2针,供试品2针的顺序进样,数据如下:[img=只进供试品和空白,690,267]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031139383447_5262_3356999_3.png!w690x267.jpg[/img]令本人百思不得其解,请各位分析解答,谢谢~最后附上检验方法(仪器为安捷伦7890B,色谱柱为安捷伦VF-17ms,砂芯衬管)[img=,690,596]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143388912_5805_3356999_3.png!w690x596.jpg[/img][img=,690,368]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143564333_3950_3356999_3.png!w690x368.jpg[/img]
高效液相色谱仪对照品的纯度要求是多少?
【求助】中检所胆固醇对照品要检测胆固醇的含量,但是中检所的胆固醇对照品是用作鉴别的,应该怎么解决这个问题?这样的对照品还能用于含量测定吗?请高手指点,谢谢!
[size=4]1.所用对照品(标准品)中检所已经发放提供(可参阅中国药典2005年版二部附录ⅩⅤG),且使用方法相同时,应使用中检所提供的现行批号对照品(标准品),并提供其标签和使用说明书,说明其批号,不应使用其他来源者;如使用方法与说明书使用方法不同(如定性对照品用作定量用、效价测定用标准品用作理化测定法定量、UV法或容量法对照品用作色谱法定量等),应采用适当方法重新标定,并提供标定方法和数据;若色谱法含量测定用对照品用作UV法或容量法,定量用对照品用作定性等,则可直接应用,不必重新标定。 2.申报临床研究时,如中检所尚无供应,为不影响注册进度,可先期与中检所接洽制备和标定,申报时提供标定报告、标签(应标明效价或含量、批号、使用效期)和使用说明书;也可与省所合作标定,申报时提供标准品或对照品研究资料,“说明其来源、理化常数、纯度、含量及其测定方法和数据”;标定有困难时,可使用国外药品管理当局或药典委员会发放的对照品(标准品)或国外制药企业的工作对照品(标准品),进行标准制订和其他基础性研究,但应提供其标签(应标明其含量)和使用说明书,能保证其量值溯源性;也可使用国外试剂公司(如sigma公司等)提供的对照品(标准品),但应提供试剂公司该批对照品(标准品)的检测报告(用作含量测定时,应有确定的含量数据),如为高纯度试剂,提供了国外试剂公司检测报告(用作含量测定时,应有确定的含量数据)时,也可使用,并应能保证其量值溯源性,但申请人应及时与中检所接洽对照品(标准品)的标定事宜,临床研究期间完成此工作。 3.直接申报生产品种,如中检所尚无供应,可参照2中要求进行,并提供相应研究资料,但申请人在标准试行期间应与中检所接洽并完成的标定事宜。 4.对照品(标准品)标定的技术要求: 4.1.创新药物 应说明对照品(标准品)原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱),提供标定方法的研究和验证资料(如与原料药质量研究项下相同,可不再提供)、含量测定数据及经统计分析得到的对照品(标准品)含量结果,并说明进行临床前药学研究、药理毒理学研究所用样品的含量是否用该批对照品(标准品)确定或可用该批对照品(标准品)进行量值溯源。 ●纯度测定方法应选用色谱法,并采用两种以上不同分离机理或不同色谱条件并经验证的色谱方法相互验证比较,同时采用二极管阵列检测器或其它适宜方法检测HPLC法的色谱峰纯度,而后根据测定结果经统计分析确定对照品(标准品)原料的纯度。 ●对于组份单一、纯度较高的药物,对照品(标准品)标定方法宜首选可进行等当量换算、精密度高、操作简便快速的容量法。可根据药物分子中所具有的官能团及其化学性质,选用不同的容量分析方法,但应符合如下条件:(1)反应按一个方向进行完全;(2)反应迅速,必要时可通过加热或加入催化剂等方法提高反应速度;(3)共存物不得干扰主药反应,或能用适当方法消除;(4)确定等当点的方法要简单、灵敏;(5)标化滴定液所用基准物质易得,并符合纯度高、组成恒定且与化学式符合、性质稳定(标定时不发生副反应)等要求。 标定方法的选择要关注如下事项:(1)供试品的取用量应满足滴定精度的要求(消耗滴定液约20ml);(2)滴定终点的判断要明确,提供滴定曲线。如选用指示剂法,应考虑其变色敏锐,并用电位法校准其终点颜色。(3)为排除因加入其它试剂而混入杂质对测定结果的影响,或便于剩余滴定法的计算,可采用“将滴定的结果用空白试验校正”的办法;(4)要给出滴定度(采用四位有效数字)的推导过程。 标定结果要根据3个以上实验室各不少于15组测定结果经统计分析,去除离群值和可疑值后的结果,并报告可信限。 ●如该药物没有可进行等当量换算并符合要求的容量法时,可采用反复纯化的原料,色谱法确定纯度后扣除有关物质、炽灼残渣、水分和挥发溶剂等后的理论含量确定为标准品含量,以此为基准进行对照品(标准品)的换代和量值传递。 ●用于抗生素微生物检定法的第一代基准标准品可参照上述方法标定,如为多组份抗生素,其组份比例应与拟上市产品组份比例一致或接近,或以其中某一组份纯品为基准标准品,但要注意标准品换代时量值传递的恒定。 ●仅用于鉴别定性的化学对照品,注重其结构确证的研究资料,纯度和含量的要求一般可适当降低。 ●杂质对照品,用作限度要求时,应提供其来源(合成路线)、结构确证的研究资料,应具备较高的纯度和含量,并提供纯度和含量的的测定结果,提供质量控制标准。 4.2其他类别药物,可参照4.1要求进行。 ●用于抗生素微生物检定法的标准品须用上市国的国家标准品或原发厂的工作标准品为基准标准品进行标定。标定时采用的原料药应符合相应要求,并提供原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱)。标定须用现行版中国药典附录收载的“抗生素微生物检定法”-三剂量法,并提供详细的方法学研究,包括检定菌和培养基的选择、剂量和剂距选择、缓冲液选择(如与质量研究项下相同,可不再提供)。每次标定结果均应照“生物检定统计法-量反应平行线测定法(3.3)”法进行可靠性测验及效价计算。按照《药品注册管理办法》,上市药品质量标准所用标准物质均须由中检所负责标定和管理,药品研发过程中,研制单位应注意及时与中检所联系标定事宜,以保证研发工作的连续性。[/size]
中药标准品等中英文名称对照货号 英文名称 中文名称019-14621 Aconitine std. 乌头碱012-14091 Albiflorin std 芍药内酯苷018-13231 Alisol B acetate,98.0% 乙酸泽泻酯B010-13431 Alisol B std 泽泻醇B015-11921 Arbutin std 熊果苷012-14611 Atractylenolide III std. 苍术内酯016-11691 Atropine sulfate std. 硫酸阿托品016-10351 Aucubin std. 桃叶珊瑚苷027-07751 Baicalein std. 黄岑素020-07741 Baicalin std. 黄岑苷022-08161 Barbaloin std. 芦荟苷022-07681 Berberine chloride std. 氯化黄连素028-12051 Bergenin std. 岩白菜内酯025-10121 Bufalin std. 蟾毒灵024-10691 Bufotalin std. 蟾毒它灵030-10611 Capillarisin std. 茵陈色原酮036-10613 Capillarisin std. 茵陈色原酮031-15141 Capsaicin std. 辣椒素032-10551 Catalpol std. 梓醇038-13711 Cinobufagin std. 华蟾毒精035-13721 Cinobufotalin std. 华蟾毒它灵036-11311 Coptisine chloride 氯化黄连碱036-14971 Corydaline std. 紫堇碱032-13731 Costunolide std. 木香烃内酯043-27611 Dehydrocorydaline nitrate std. 硝酸脱氢紫堇碱040-21881 Dehydrocostuslactone std. 脱氢广木香内酯044-25321 Dihydrocapsaicin std. 二氢辣椒素042-18911 Dimethylesculetin std. 二甲基七叶树内酯052-04921 Ergosterol std. 麦角甾醇056-05161 β-Eudesmol std. β-桉油醇050-04601 Evodiamine std. 吴茱萸碱070-02241 Geniposide std. 京尼平苷078-02301 Geniposidic acid std. 京尼平苷酸077-02871 6-Gingerol std. 6-姜辣醇079-02191 Ginsenoside Rb1 std. 人参皂苷Rb1078-03261 Ginsenoside Rc 人参皂苷Rc072-03301 Ginsenoside Rd std. 人参皂苷Rd075-03271 Ginsenoside Re std. 人参皂苷Re072-02201 Ginsenoside Rg1 std. 人参皂苷Rg1071-02271 Glycyrrhizin std. 甘草甜素089-04951 Honokiol std. 和厚朴酚081-06851 (E)-10-Hydroxy-2-decenoic acid std. (E)-10-羟基-2-癸烯酸084-07061 Hypaconitine 次乌头碱099-03651 Isofraxidine std. 125-03361 Liquiritin std. 甘草甙125-03621 Loganin std. 番木鳖苷137-09081 Magnolol std. 厚朴酚130-12261 Mesaconitine 中乌头碱140-06211 Naringin std. 柚皮甙151-01801 Osthole std. 甲氧基欧芹酚150-01511 Oxymatrine std. 氧化苦参碱167-11711 Paeoniflorin std. 芍药苷163-11713 Paeoniflorin std. 芍药苷169-12871 Paeonol std. 牡丹酚166-17641 Palmatine chloride std. 氯化巴马亭164-14091 (+)-phyllodulcin std. (+)-叶甜素162-14071 Puerarin std. 葛根素184-00951 Resibufogenin std. 残余蟾蜍配基183-00921 Rutaecarpine std. 吴茱萸次碱190-08411 Saikosaponin a std. 柴胡皂甙a194-10021 Saikosaponin b2 std. 柴胡皂甙b2197-08421 Saikosaponin c std. 柴胡皂甙c194-08431 Saikosaponin d std. 柴胡皂甙d192-10441 Schizandrin std. 五味子素197-09261 Scopolamine hydrobromide, 99.0% 氢溴酸莨菪胺190-08531 Sennoside A std. 番泻苷A194-09271 Sennoside B, 96% 番泻苷B199-11811 Sennoside B std. 番泻苷B191-08681 Shikonin std. 紫草素193-09361 Sinomenine std. 青藤碱197-08541 Swertiamarin std. 獐牙菜苦苷231-00811 Wogonin std. 汉黄岑素
请问各位老师做异丙醇和蔗糖红外鉴别用的什么对照品,中检院的对照品没说可以用于红外鉴别能用吗
请问做HPLC 可以用USP 纯度为91.7%的对照品么?谢谢
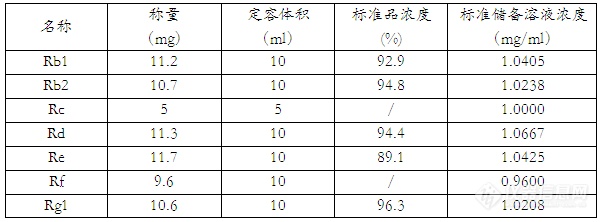
一、实验目的及原理:参照药典中人参皂苷的检测,使用超高效液相色谱,对于人参中7种皂苷成分进行检测,大大提高了检测速度,并拓展了检测的皂苷种类。人参中的皂苷成分通过超声波萃取,高校液相色谱柱进行分离,采用紫外检测器进行检测,外标法定量。二、实验仪器及试剂:仪器:UPLC(waters H-Class),检测器:TUV紫外检测器、超声波仪、超纯水机、离心机、涡旋振荡仪、50ml离心管、20ml胖肚移液管试剂:人参皂苷标准品Rb1、Rb2、Rd、Re、Rf、Rg1购自食品药品检定院, 人参皂苷标准品Rc购自百灵威、乙腈(HPLC)、甲醇(HPLC)三、色谱条件:色谱柱:BEHC18 2.1mm×50 mm×1.7um 柱温:35℃ 样品温度:25℃进样体积:5µL 流速:0.4ml/min 波长:203nm 采样速率:20点/秒流动相配比:时间水乙腈曲线类型08218/1082186185050618.1821862082186四、实验步骤4.1、标准储备溶液配制:称取人参皂苷标准品,用甲醇溶解定容,具体见下表:http://ng1.17img.cn/bbsfiles/images/2012/10/201210241734_399095_1854832_3.jpg4.2、样品处理称取样品约0.4g(精确到0.0001g)于50ml离心管中,加入20.0ml超纯水,涡旋15s,超声波萃取30min,5000转/min离心5min,过0.22um水膜,待测。五、计算5.1样品中人参皂苷浓度的计算: X=C*100/m式中:X——样品中人参皂苷的浓度(mg/g) m——样品称量(g) C——待测溶液中人参皂苷的浓度(ug/mL)六、标准曲线与线性相关系数取标准储备溶液混合,用水稀释至约5、10、20、50、100ug/ml,进行2次平行测定,得到相应的峰面积平均值。以浓度为横坐标,以峰面积为纵坐标,绘制标准曲线,结果如下:http://ng1.17img.cn/bbsfiles/images/2012/10/201210241728_399091_1854832_3.jpg七、样品测定及回收率实验(以6年红参体为样本)http://ng1.17img.cn/bbsfiles/images/2012/10/201210241730_399092_1854832_3.jpg八、对照与样品测定的色谱图http://ng1.17img.cn/bbsfiles/images/2012/10/201210241731_399093_1854832_3.jpg九、结果与讨论本实验尝试多个色谱条件, Re、Rg1组分性质非常接近较难分离,选择梯度洗脱的方式加快其他组分的分离,相对于传统液相方法需要至少一个小时的进样时间,UPLC的分离时间在20min,具有较大的优势。







 我要推广仪器
我要推广仪器
 下载APP
下载APP