现在我们公司增加辅料乙醇和醋酸钠的红外检测,需要购买红外乙醇对照品和醋酸钠对照品。请各位大侠提供除了中检所外,能够在一个星期内买到的厂家。谢谢大家了!!!!
色甘酸钠发现的故事最近研究色甘酸钠发现的故事,这里涉及的科学家个人性格,药物研发模式,先导物质量,锲而不舍的毅力等因素可以给今天的药物工作者以很多启示。色甘酸钠是由一位名叫Roger Altounyan的医生所发现。此人天生喜欢冒险,二战时谎报年龄(因为年纪太小)加入英国空军成为一名轰炸机飞行员。因其出色的飞行技术后来成为飞行教官和教官的教官,并发明了一种非常危险的夜间低空飞行技术。他的冒险家性格后来成为其发现色甘酸钠的关键因素。二战后他继承父业去剑桥读医学院(他父亲和祖父均为医生)但毕业后找不到医生位置所以加入一个叫做Bengers的制药公司(后卖给赛诺菲和阿斯列康),研究抗哮喘药。当时哮喘药的模型是组胺诱导豚鼠哮喘模型,但作为哮喘患者的Altounyan发现抗组胺药物不能缓解自己的哮喘,所以认为这个动物模型不好,用今天的话来说就是转化性较差。那什么模型好呢?他认为他自己是最好的模型。他发现自己吸入捣碎的豚鼠毛可以诱发严重的哮喘,在1957-1965的8年间他共吸入豚鼠毛1000多次,曾几度差点丧命。有了好的模型到哪找先导物呢?他是土耳其后裔,知道中东有一种叫做khellin的草药可以治哮喘。他和化学家合作从这个草药里寻找有效成分。开始几年几乎没找到什么有用的东西。1961年公司决定停止这个项目。多数人在自已自愿哮喘5年后都会和公司一起放弃了,但这老兄很轴,还要继续。因为公司不再支持这个项目所以所有的新化合物都不再做安全性研究,他可以继续自测新化合物但生死无论。1963年,终于有一个新化合物可以100%抵制豚鼠毛在Altounyan本人诱发的哮喘。所以公司把这个化合物送进临床给一位哮喘病人使用。病人连续吸入这个化合物60小时发现丁点儿用没有。难道是豚鼠毛诱导的哮喘和天然哮喘不一样?Altounyan自己又试了一次这个化合物(又自己诱发了一次哮喘!)发现也无效。后来和化学家一讨论基本认定虽然两批药品是按同一路线合成但给病人那批更纯。在回去找他老先生自己用的那批糙货发现里面有少量的杂质,即后来的色甘酸钠。色甘酸钠在组胺诱导豚鼠动物模型无效,所以人体模型是必须的。色甘酸钠成为第一个专门针对哮喘的药物,直至今天还在临床上使用。Altounyan在此后的20年间还继续用豚鼠毛自己诱发哮喘,寻找新的抗哮喘药物,直至1987年去世,但遗憾的是没有任何新的发现。色甘酸钠的发现有很多我们可以借鉴的东西。药物发现的有些复杂步骤无法还原成更便宜,简单的测试。这是一个非常关键的理念,基因-蛋白-细胞-动物模型-病人之间的转化在很多疾病无法实现,所以不管体外活性怎么便宜,构效关系如何清晰,都对发现新药没有帮助。这样的数据是自欺欺人,只是智力游戏。人体和动物显然有很大区别,组胺可以诱导动物哮喘但抗组胺药对哮喘病人无效而色甘酸钠对哮喘病人有效但在动物模型无效。Altounyan本人的冒险和牺牲精神是色甘酸钠发新的根本因素,但是如果没有他百折不挠的毅力和胆大心细的素质,色甘酸钠也不会被发现。色甘酸钠的分子结构较为怪异,按今天的眼光看类药性很差,我估计如果这个化合物成为HTS的苗头化合物很多团队不会跟踪优化它。类药性是药物化学最傲慢的概念,应该禁止使用。我们根本就不知道药物应该长什么样。最后,运气起了很重要的作用。色甘酸钠发现后的20年Altounyan继续用自己筛选新药但并无斩获。色甘酸钠是药物发现史的一个重要事件,Altounyan为此做出了巨大牺牲,他晚年肺病严重。但苦心人,天不负,色甘酸钠减少了无数哮喘病人的痛苦,50年后的今天依然在临床中使用。
样品为复方维生素,其中一项是核黄素磷酸钠,没找到核黄素磷酸钠的对照品,故用的核黄素对照品样品制备:先用水溶,然后用流动相稀释,流动相弱酸性做出的结果比标示量低了很多啊用核黄素对照品代替核黄素磷酸钠对照品,请问结果可信吗?
高效液相色普法测定甘草酸苷含量,甘草酸苷对照品峰面积与之前相比较偏高,导致含量偏高,请问有可能是哪些原因?
请问有谁做过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测辛酸钠有关物质的实验吗?您做的对照溶液的信噪比大概是多少?谢谢!请问辛酸钠有关物质检查法 中 供试品溶液中的溶剂是什么?每次试验前需要先进一针溶剂吗?是不是要根据信噪比的要求来调节纵坐标的范围 使对照溶液的主成分峰的峰高约为满量程的10-25% ?
请问一下各位:车前醚苷对照品哪里有卖呢?
对照品甘草苷 后面出杂峰 是怎么回事[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221432219383_6429_3461163_3.jpeg[/img]
http://ng1.17img.cn/bbsfiles/images/2012/09/201209272130_393425_2255248_3.gifHPLC测定木犀草苷对照品为什么峰前面有个小峰????
配制两个黄芩苷对照品溶液第一个是用50%的甲醇溶解,我感觉这个好难溶,超声了也不见得全部溶解,如果全部溶解配制出来的应该是澄清的吧?该如何处理好呢??第二个要用减压干燥器60度干燥4小时再配制,我觉得涂了凡士林在60度会溶了吧,就不能减压了,我可以直接打开对照品瓶盖直接在烘箱里烘4小时不?这样做会有很大的影响吗?
大家都用那些容积配置过黄芩苷对照品啊?怎么我们用稀乙醇溶液溶解的时候不易溶解啊,超声半小时还会有很多不容物?
[em06] 我要用玻璃酸钠、右旋糖酐70、羟丙甲纤维素这三种对照品,可是没有什么好的溶解方法,对结果影响很大。请问哪位高手可以赐教?谢谢!!!
我在做陈皮含量的时候,橙皮苷对照品用甲醇不溶解,请问谁可以指点一下?
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7681612594 七氟醚杂质C Sevoflurane Related Compound C 对照品/标准品1612572 七氟醚杂质 B Sevoflurane Related Compound B 对照品/标准品1612550 七氟醚杂质 A Sevoflurane Related Compound A 对照品/标准品1612540 七氟醚 Sevoflurane 对照品/标准品1612539 盐酸舍曲林 Sertraline Hydrochloride 对照品/标准品1612528 盐酸舍曲林杂质A Sertraline Hydrochloride Related Compound A 对照品/标准品1612517 盐酸舍曲林消旋体混合物 Sertraline Hydrochloride Racemic Mixture 对照品/标准品1612506 L-丝氨酸 L-Serine 对照品/标准品1612426 芝麻油杂质B Sesame Oil Related Compound B 对照品/标准品1612415 芝麻油杂质A Sesame Oil Related Compound A 对照品/标准品1612404 芝麻油 Sesame Oil 对照品/标准品1612029 番泻苷 B Sennoside B 对照品/标准品1612018 番泻苷 A Sennoside A 对照品/标准品1612007 番泻苷 Sennosides 对照品/标准品1611955 硒 蛋氨酸 Selenomethionine 对照品/标准品1611900 盐酸司来吉兰 Selegiline Hydrochloride 对照品/标准品1611004 司可巴比妥 CII Secobarbital CII 对照品/标准品1610090 东莨菪亭 Scopoletin 对照品/标准品1610001 氢溴酸东莨菪碱 Scopolamine Hydrobromide 对照品/标准品1609831 沙奎那韦杂质A Saquinavir Related Compound A 对照品/标准品1609829 甲磺酸沙奎那韦 Saquinavir Mesylate 对照品/标准品1609807 双水杨酯 Salsalate 对照品/标准品1609625 沙美特罗杂质B Salmeterol Related Compound B 对照品/标准品1609614 沙美特罗杂质A Salmeterol Related Compound A 对照品/标准品1609603 昔美酸沙美特罗 Salmeterol Xinafoate 对照品/标准品1609501 水杨酸片 Salicylic Acid Tablets 对照品/标准品1609024 水杨酸杂质B Salicylic Acid Related Compound B 对照品/标准品1609013 水杨酸杂质A Salicylic Acid Related Compound A 对照品/标准品1609002 水杨酸 Salicylic Acid 对照品/标准品1608000 水杨酰胺 Salicylamide 对照品/标准品1607506 连翘粉状贯叶提取物 Powdered St. John's Wort Extract 对照品/标准品1607040 糖精钠 Saccharin Sodium 对照品/标准品1607029 糖精钙 Saccharin Calcium 对照品/标准品1607007 糖精 Saccharin 对照品/标准品1606503 芦丁 Rutin 对照品/标准品1606208 硝酚胂酸 Roxarsone 对照品/标准品1605523 罗哌卡因杂质B Ropivacaine Related Compound B 对照品/标准品1605512 罗哌卡因杂质A Ropivacaine Related Compound A 对照品/标准品1605500 盐酸罗哌卡因 Ropivacaine Hydrochloride 对照品/标准品1604916 罗库溴铵合剂峰的识别 Rocuronium Peak Identification Mixture 对照品/标准品1604905 罗库溴铵 Rocuronium Bromide 对照品/标准品1604870 利凡斯的明杂质B Rivastigmine Related Compound B 对照品/标准品1604869 利凡斯的明杂质A Rivastigmine Related Compound A 对照品/标准品1604814 利托那韦杂质混合物 Ritonavir Related Compounds Mixture 对照品/标准品1604803 利托那韦 Ritonavir 对照品/标准品1604701 盐酸利托君 Ritodrine Hydrochloride 对照品/标准品1604665 利培酮系统适用性试验用混合物 Risperidone System Suitability Mixture 对照品/标准品1604654 利培酮 Risperidone 对照品/标准品1604643 利塞膦酸杂质C Risedronate Related Compound C 对照品/标准品1604632 利塞膦酸杂质B Risedronate Related Compound B 对照品/标准品1604621 利塞膦酸杂质A Risedronate Related Compound A 对照品/标准品1604610 利塞膦酸钠 Risedronate Sodium 对照品/标准品1604600 利美索龙 Rimexolone 对照品/标准品1604508 盐酸金刚乙胺 Rimantadine Hydrochloride 对照品/标准品1604348 利鲁唑杂质A Riluzole Related Compound A 对照品/标准品1604337 利鲁唑 Riluzole 对照品/标准品1604202 醌式利福平 Rifampin Quinone 对照品/标准品1604009 利福平 Rifampin 对照品/标准品1603800 利福布丁 Rifabutin 对照品/标准品1603108 核糖 Ribose 对照品/标准品1603006 维生素B2 Riboflavin (Vitamin B2) 对照品/标准品1602706 利巴韦林 Ribavirin 对照品/标准品1602003 间苯二酚 Resorcinol 对照品/标准品1601849 二类残留溶剂-二甲苯 Residual Solvent Class 2 - Xylenes 对照品/标准品1601827 二类残留溶剂-三氯乙烯 Residual Solvent Class 2 - Trichloroethylene 对照品/标准品1601805 二类残留溶剂-甲苯 Residual Solvent Class 2 - Toluene 对照品/标准品1601780 二类残留溶剂-四氢萘 Residual Solvent Class 2 - Tetralin 对照品/标准品1601770 二类残留溶剂-四氢呋喃 Residual Solvent Class 2 - Tetrahydrofuran 对照品/标准品1601769 二类残留溶剂-二氧噻吩烷 Residual Solvent Class 2 - Sulfolane 对照品/标准品1601747 二类残留溶剂-吡啶 Residual Solvent Class 2 - Pyridine 对照品/标准品1601725 二类残留溶剂-硝基甲烷 Residual Solvent Class 2 - Nitromethane 对照品/标准品1601703 二类残留溶剂-N-甲基吡咯烷酮 Residual Solvent Class 2 - N-Methylpyrrolidone 对照品/标准品1601689 二类残留溶剂-甲基环己烷 Residual Solvent Class 2 - Methylcyclohexane 对照品/标准品1601667 二类残留溶剂-甲基丁基酮 Residual Solvent Class 2 - Methylbutylketone 对照品/标准品1601645 二类残留溶剂- 2-甲氧基乙醇 Residual Solvent Class 2 - 2-Methoxyethanol 对照品/标准品1601623 二类残留溶剂-甲醇 Residual Solvent Class 2 - Methanol 对照品/标准品1601601 二类残留溶剂-己烷 Residual Solvent Class 2 - Hexane 对照品/标准品1601587 二类残留溶剂-甲酰胺 Residual Solvent Class 2 - Formam
给位大神求帮我分析一下顶空进样测苯,苯对照品的溶剂是90%乙醇,加了10mg 的无水碳酸钠,三针对照总会有一针峰面积较小(只有一半),是什么原因?
求助:中药口服液质量标准制定的一些问题 第一次做新药,是一个中药复方口服液,主要成分中含有氢溴酸东蒗菪碱,质量标准制定如下: 5.1.1色谱条件 色谱柱:Promosil C18(250×4.6mm,5洀,博纳艾杰尔公司);流动相:0.07mol/L磷酸钠溶液 (用磷酸调 pH值至6.0,含0.0175mol/L十二烷基硫酸钠)-乙腈 (2:1);流速:0.8ml/min;检测波长:216nm;柱温:25℃;进样量:10ul。理论塔板数按氢溴酸东莨菪碱峰计算应不低于10000。 5.1.2 供试品和对照品溶液的制备 5.1.2.1 对照品溶液 精密称取氢溴酸东莨菪碱10.18mg,置10ml容量瓶中,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)使溶解,并稀释至刻度,摇匀,制成每1ml含1.018mg的溶液(东莨菪碱重量=氢溴酸东莨菪碱/1.445)。 5.1.2.2 供试品溶液 精密量取本品溶液5ml,置锥形瓶中,加入2mol/L盐酸溶液30ml,超声处理(功率250W,频率40kHz)30分钟,放冷,滤过,滤渣和滤器用 2mol/L盐酸溶液分数次洗涤,合并滤液和洗液,用浓氨试液调节pH值至9,用三氯甲烷振摇提取4次,每次30ml,合并三氯甲烷液,回收容剂至干,残渣用0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)溶解并移至5ml量瓶中,稀释至刻度,摇匀,即得。 5.1.3 阴性对照品溶液的制备 按处方比例及工艺,制备缺洋金花的阴性样品,再按供试品溶液的制备方法制备不含洋金花的阴性样品溶液。 5.1.4 测定方法 精密吸取上述对照品溶液与供试品溶液各10u1,分别注入液相色谱仪(自动进样仪),测定峰面积,以外标法计算含量。 5.1.5 系统适用性试验 分别吸取对照品溶液、供试品溶液及阴性品溶液10ul注入高效液相色谱仪,阴性对照品在氢溴酸东莨菪碱峰位置处无吸收峰,即其它成分对测定无干扰,氢溴酸东莨菪碱峰与其它组分峰基线分离良好。 5.1.6 线性试验 精密称取氢溴酸东莨菪碱对照品10.16mg,置25ml量瓶中,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)溶解并稀释至刻度,摇匀。精密吸取0.5、1.0、1.5、2.0、2.5、3.0ml,分别置10ml量瓶中,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)溶解并稀释至刻度,摇匀。按上述色谱条件测定,分别进样10ul,测得峰面积积分值。以对照品的浓度为横坐标,峰面积积分值为纵坐标,绘制标准曲线,得回归方程为:A=1.3×107C-9243.77,相关系数r:=0.9997。表明进样量在0.2032~1.2192ug范围内呈良好线性关系。 5.1.7 稳定性试验 精密量取供试品溶液5ml,按供试品(批号:2009010501)溶液制备方法制成供试品溶液,间隔一定时间(0,4,8,12,24,48h)重复进样,每次进样10ul,测得峰面积的RSD为1.78%,表明该溶液在48小时基本稳定。 5.1.8 精密度试验 精密吸取上述对照品溶液10ul,重复进样6次,测定东莨菪碱的峰面积,结果 RSD%为0.58%(n=6)。表明本方法精密度良好。 5.1.9 重复性试验 精密量取供试品溶液(批号:2009010501)5ml,按供试品溶液制备方法平行制得6份供试品溶液,分别测定东莨菪碱的含量,结果RSD为1.42%(n=6)。表明本方法有较好的重复性 。 5.1.10 加样回收率试验 5.1.10.1 对照品溶液的制备 精密称取氢溴酸东莨菪碱对照品约7.8mg,置100ml的容量瓶内,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)使溶解,并稀释至刻度,即得。 5.1.10.2 采用加样回收率测定方法,精密量取已知含量的样品2.5ml,精密加入上述对照品溶液2.5ml/2ml/3ml,按供试品溶液制备方法平行制得6份供试品溶液,依法测定,结果平均回收率为106.7%,RSD为1.42%。 回收率=(测的东莨菪碱含量—样品中东莨菪碱含量)/加入东莨菪碱量 因为是第一次作含量测定,有许多不明白之处,中药质量标制定准指导原则也说得不清不楚,许多都是按照自己理解摸索来的。 通过测定样品的平均含量为0.077mg/ml。线性试验是根据样品的平均含量,以样品的平均含量为中间值,制定的线性取样范围。 根据加样回收中对照品加入量与样品含量之和在线性范围的原则,按照80%,100%,120%的原则加入对照品,但是结果很不理想,加样回收率超过了200%,不知道是什么方面的原因,很着急,请大家帮忙分析一下,找出有错误的地方,我马上更改。

中药制剂中木瓜的薄层鉴别:供试品与对照药材处相应的点颜色不同是为什么?狗脊的薄层鉴别也是一样,提取时处理方法是一样的图1前三供试品(绿色),中二木瓜对照药材(蓝色),后二阴性对照图2前二供试品(亮的点蓝色),中二狗脊对照药材(绿色),后二阴性对照不知道该怎么办,诚请各位大师解答,谢谢[img=,690,459]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329352099_2969_1816508_3.jpg!w690x459.jpg[/img][img=,380,362]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329533546_2463_1816508_3.jpg!w380x362.jpg[/img]
[color=#444444]HPLC测定人参皂苷Ra1对照品含量以色谱纯乙腈-水为流动相,为什么所得结果只有前五分钟的溶剂峰,却没有想要的保留时间为16分钟左右的峰呢?[/color]
各位大侠,谁那里有硝酸钠水溶液与波美度的对照表啊?有的话请发份给我,谢谢。急。。。。
椰油酰甘氨酸钠测定盐含量的时候颜色变化不明显该怎么办?为什么加入硝酸银的时候会有很多白色物质,而且盐含量COA上面明明很低。求助各位大神。
椰油酰甘氨酸钠中的盐含量测试出了滴定方法,还有什么方法可以测试吗,滴定中颜色变化太不明显,想看看其他办法,请大神指点一二。
因为刚接触药品检验,在做核黄素磷酸钠的游离磷酸的测定的时候,做了几次都不合格。 按照标准要求进行对照及样品配制,对照品为蓝色的澄清液体,但是样品很浑浊,土黄色。在730nm处进行测定,结果不符合规定。 因为样品很浑浊,所以吸收值很大(2.0),大过了对照品; 我们把样品用0.45的滤膜过滤,吸收值(0.35)低于对照,但是我们怀疑过滤的影响太大,我们就再次过滤样品,吸收值又降低了一半(0.13). 请各位大侠帮帮忙啊!!!
草乌薄层喷稀碘化铋钾试液后三种对照品都显什么颜色
按药典测定甘油磷酸钠中磷含量,用钼酸铵,对苯二酚,乙酸钠,配制好后,显蓝色,720nm波长检测,吸光度一直降低,原来0.629,一个小时后,0.518,第二天,颜色是淡淡的蓝色了,这是为什么?
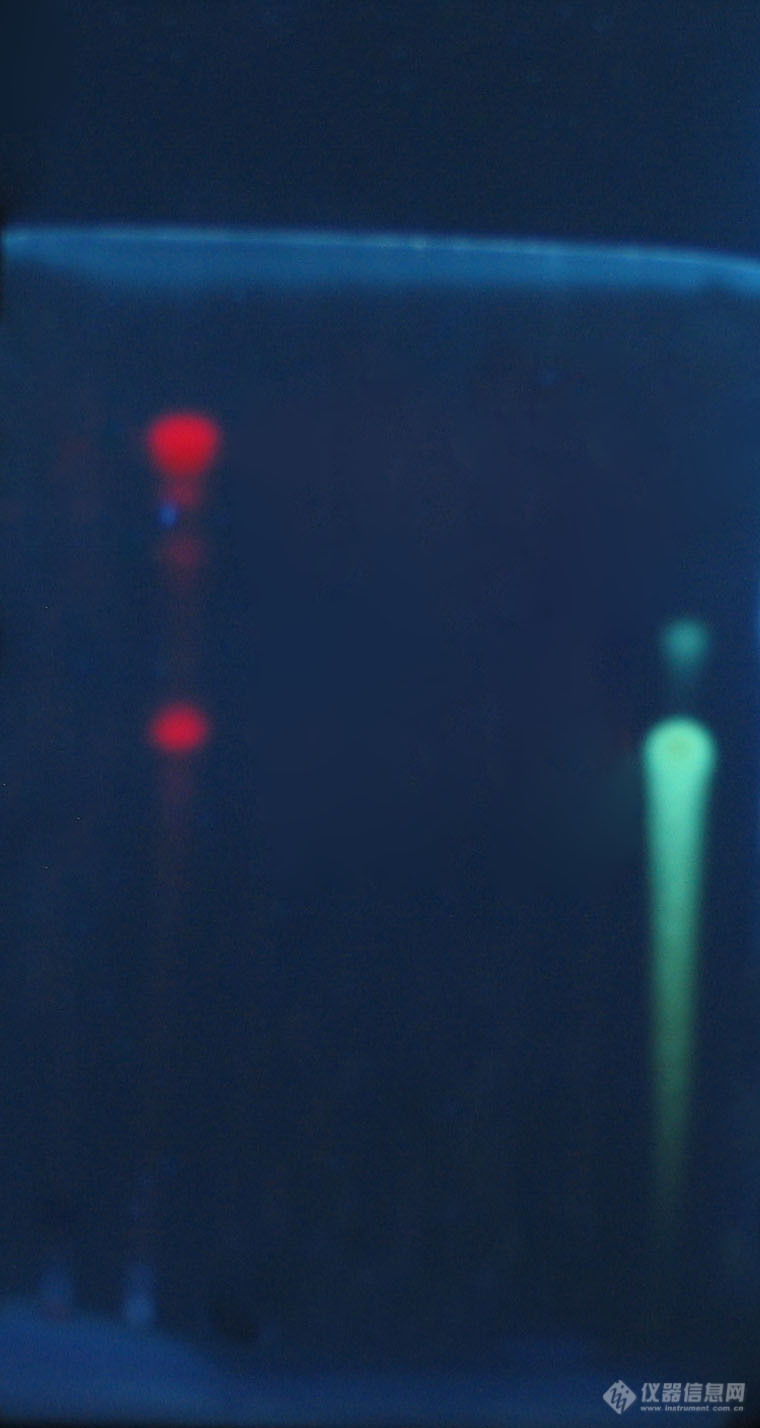
如果薄层色谱中,供试品和对照品在相应的位置上显示不同颜色的斑点说明什么?另,对照品显示两个距离很近颜色相同的点。[img=,690,1296]https://ng1.17img.cn/bbsfiles/images/2019/09/201909091310514179_9415_1786659_3.jpg!w690x1296.jpg[/img]
做氯霉素滴眼液有关物质:流动相:以含0.1%庚烷磺酸钠溶液(取0.1%庚烷磺酸钠溶液500ml与二甲基酰胺5ml、冰醋酸0.5ml,混匀)-乙腈(72:28)(药典为75:25)为流动相 柱子:Hypersil Bos C18 5μm尺寸:4.6×250mm庚烷磺酸钠 天津傲然精细化工研究所对硝基苯甲醛 Fluka Chemie GmbH CH-9471 Buchs结果对硝基苯甲醛一项对照品与样品的保留时间相差1.5min,氯霉素二醇物一致.请问与什么因素有关?与改变流动相比例有关系吗?流动相中二甲基甲酰胺、冰醋酸起什么作用?
对照品溶液与供试品溶液需使用相同的顶空条件,因为样品瓶中有气-液或气-固两相,甚至气-液-固三相共存。顶空气体中各组分的含量既与其本身的挥发性有关,又与样品基质有关。特别是那些在样品基质中溶解度大(分配系数大)的组分,“基质效应”更为明显。这是顶空进样的一大特点,即顶空气体的组成与原样品中的组成不同,这对定量分析的影响尤为严重。因此,对照品不能仅用待测物的标准品配制,还必须有与原样品相同或相似的基质,否则,定量误差将会很大。实际应用中一些消除或减少基质效应的方法:a、利用盐析作用即在水溶液中加入无机盐(如过饱和硫酸钠溶液)来改变挥发性组分的分配系数;b、在有机溶液中加入水相,这可以减小有机物在有机溶剂中的溶解度,增大其在顶空气体中的含量;c、调节溶液的pH 对于碱和酸,通过控制pH可使其解离度改变,或使其中待测物的挥发性变得更大,从而有利于分析;d、降低样品浓度也是减小基质效应的常用方法,但其代价是减低了灵敏度;e、固体样品中挥发物的扩散速度很慢,往往需要很长时间才能达到平衡。尽量采样全溶的样品溶液,有利于缩短平衡时间;f、其他消除基质效应的技术,如全挥发技术等
我是做中药检验的,以前用的对照品溶液都经0.45滤膜过滤后使用,还做过某两种对照品溶液的测试,峰面积差异不大。但最近我使用黄芩苷对照品时发现检品含量总是很高,反复测试才找到原因,过滤后对照品的峰面积为:3249725.000,没过滤的对照品峰面积为:4933873.000,差异很大,不知道各位朋友使用对照品溶液时是否也经过0.45滤膜过滤?有没有文件规定对照品溶液能否过滤?
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。
在薄层色谱定性时,对照品、供试品和对照药材在相同的问题显相同颜色的斑点即可。对照品和对照药材定性有何区别?为什么两个要同时做呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP