延胡索薄层样品比对照药材多了一个点,怎么回事?
在薄层色谱定性时,对照品、供试品和对照药材在相同的问题显相同颜色的斑点即可。对照品和对照药材定性有何区别?为什么两个要同时做呢?

黄连上清片高温灭菌后,黄连对照药材薄层鉴别的点变红是怎么回事?[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2019/07/201907101138123673_5943_1782490_3.jpg!w690x388.jpg[/img]
研究中药复方制剂中各味药材的鉴别药典中鉴别乌梅药材,选择“熊果酸”为对照乌梅含有熊果酸,大枣也含有熊果酸,请问那我做制剂中乌梅药材鉴别时,是不是就不能以熊果酸作对照了,因为怕大枣会有干扰?这种情况,乌梅该选择什么组分作为对照?实验结果显示,乌梅阴性并没有干扰,我能以“熊果酸”作为对照鉴别乌梅吗,虽然明知大枣也含熊果酸。乌梅阴性没有干扰,会不会是点样量少的问题?http://ng1.17img.cn/bbsfiles/images/2012/11/201211010017_400582_1872149_3.jpg1. 熊果酸;2-4. 样品;5. 阴性

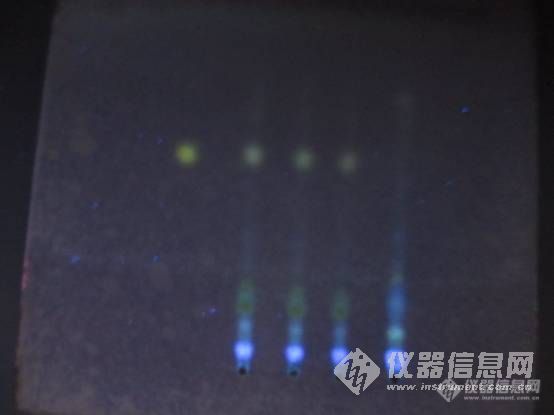
请各位老师帮忙看看,不知荆芥对照药材(右边一个为对照药材,左边三个为制剂样品——含荆芥)本身就这样,还是我做的不好,斑点不清晰。多谢![img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907171127582678_7811_1825519_3.jpg!w690x517.jpg[/img]
白芷的荧光鉴别白芷是与肉桂、红花、川乌、草乌、荆芥、防风、干姜、金银花、当归、三棱、莪术混提,用水煎煮提取2次,每次2小时,成品中有白芷的薄层鉴别,与白芷对照药材在紫外下观察荧光。我已经做了多批,都看不到荧光斑点,很困惑,请有经验的老师帮忙指导。
漏芦 本品为菊科植物祁州漏芦Rhaponticum uniflorum (L.)DC.的干燥根。春、秋二季采挖,除去须根和泥沙,晒干。 性状| 本品呈圆锥形或扁片块状,多扭曲,长短不一,直径1~2.5cm。表面暗棕色、灰褐色或黑褐色,粗糙,具纵沟及菱形的网状裂隙。外层易剥落,根头部膨大,有残茎和鳞片状叶基,顶端有灰白色绒毛。体轻,质脆,易折断,断面不整齐,灰黄色,有裂隙,中心有的呈星状裂隙,灰黑色或棕黑色。气特异,味微苦。 鉴别| (1)本品横切面:表皮常已脱落,后生皮层为数层至20余层棕色细胞,壁稍厚,木化及木栓化。韧皮部较宽广,射线宽。形成层成环。木质部导管较多,大型导管群常与小型导管群相间排列;木射线常有径向裂隙,中央有时呈星状裂隙,其周围的细胞壁木栓化。薄壁组织中有分泌管分布,内含红棕色分泌物。 粉末棕色。网纹导管和具缘纹孔导管较多,直径约至133μm。分泌管长条状,直径24~68μm,内含红棕色分泌物。根头部非腺毛细胞甚长,木化,长0.5~4μm,直径20~30μm。后生皮层细胞类方形或长方形,壁稍厚,红棕色,木化和木栓化。 (2)取本品粉末1g,加甲醇20ml,超声处理20分钟,滤过,滤液蒸干,残渣加乙酸乙酯1ml使溶解,作为供试品溶液。另取漏芦对照药材1g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以环己烷-丁酮(4:1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。 检查| [/font] 水分 不得过15.0%(通则0832第二法)。 酸不溶性灰分 不得过5.0% (通则2302)。 【浸出物】 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于8.0%。 【含量测定】 照高效液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(31:69)为流动相,待蜕皮甾酮色谱峰出峰后,用甲醇洗脱6分钟;检测波长为247nm。理论板数按β-蜕皮甾酮峰计算应不低于6000。 对照品溶液的制备 取β-蜕皮甾酮对照品适量,精密称定,加甲醇制成每1ml含20μg的溶液,即得。 供试品溶液的制备 取本品粉末(过三号筛)约1g,精密称定,精密加入30%甲醇20ml,称定重量,加热回流1小时,放冷,再称定重量,用30%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。 本品按干燥品计算,含蜕皮甾酮(C27H44O7)不得少于0.040%。 其他| 【炮制】 除去杂质,洗净,润透,切厚片,晒干。 本品呈类圆形或不规则的厚片。外表皮暗棕色至黑褐色,粗糙,有网状裂纹。切面黄白色至灰黄色,有放射状裂隙。气特异,味微苦。 【检查】 酸不溶性灰分 同药材,不得过4.0%。 【浸出物】 同药材,不得少于6.0%。 【鉴别】(除横切面外) 【[b]检查】(水分) 【含量测定】 同药材。 【性味与归经】 苦,寒。归胃经。 【功能与主治】 清热解毒,消痈,下乳,舒筋通脉。用于乳痈肿痛,痈疽发背,瘰疬疮毒,乳汁不通,湿痹拘挛。 【用法与用量】 5~9g。 【注意】 孕妇慎用。 【贮藏】 置通风干燥处。

中药制剂中木瓜的薄层鉴别:供试品与对照药材处相应的点颜色不同是为什么?狗脊的薄层鉴别也是一样,提取时处理方法是一样的图1前三供试品(绿色),中二木瓜对照药材(蓝色),后二阴性对照图2前二供试品(亮的点蓝色),中二狗脊对照药材(绿色),后二阴性对照不知道该怎么办,诚请各位大师解答,谢谢[img=,690,459]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329352099_2969_1816508_3.jpg!w690x459.jpg[/img][img=,380,362]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329533546_2463_1816508_3.jpg!w380x362.jpg[/img]
西青果的薄层图?按药典方法提取展开,对照药材也没有斑点。是否有其他可行的方法?

作者:曾中强(怀化市中医院药剂科,湖南怀化,418000)摘要: 目的:建立高效液相色谱法测定益母草药材及其颗粒制剂中水苏碱含量的方法.方法:色谱柱为Diamonsil C18(4.6mm×250mm);流动相为甲醇-0.05mol/L磷酸二氢钾溶液(70:30);检测波长210 nm,柱温25℃,流速1.0mL/min.结果:益母草药材及其制剂中水苏碱的保留时间均为15.1 min,色谱峰分离良好,对照品水苏碱线性范围为1.01~30.39 ug/mL(R2=O.9997,n=5),方法回收率为100.1%,RSD为1.03%。结论:该法可用于益母草流浸膏中及其颗粒制剂中水苏碱的含量测定,可以用来对提取物及相关制剂进行质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208071358_382237_1609970_3.jpg
槲寄生 Hujisheng 本品为桑寄生科植物槲寄生Viscum coloratum(Komar.)Nakai的干燥带叶茎枝。冬季至次春采割,除去粗茎,切段,干燥,或蒸后干燥。 性状| 本品茎枝呈圆柱形,2~5叉状分枝,长约30cm,直径0.3~1cm;表面黄绿色、金黄色或黄棕色,有纵皱纹;节膨大,节上有分枝或枝痕;体轻,质脆,易折断,断面不平坦,皮部黄色,木部色较浅,射线放射状,髓部常偏向一边。叶对生于枝梢,易脱落,无柄;叶片呈长椭圆状披针形,长2~7cm,宽0.5~1.5cm;先端純圆,基部楔形,全缘;表面黄绿色,有细皱纹,主脉5出,中间3条明显;革质。气微,味微苦,嚼之有黏性。 鉴别| (1)本品茎横切面:表皮细胞长方形,外被黄绿色角质层,厚19~80μm。皮层较宽广,纤维数十个成束,微木化;老茎石细胞甚多,单个散在或数个成群,韧皮部较窄,老茎散有石细胞。形成层不明显。木质部散有纤维束;导管周围纤维甚多,并有少数异形细胞。髓明显。薄壁细胞含草酸钙簇晶和少数方晶。 本品茎粉末淡黄色。表皮碎片黄绿色,细胞类长方形,可见气孔。纤维成束,直径10~34μm,壁较厚,略成波状,微木化。异形细胞形状不规则,壁较厚,微木化,胞腔大。草酸钙簇晶直径17~45μm;方晶较少,直径8~30μm。石细胞类方形、类多角形或不规则形,直径42~102μm。 (2)取本品粉末1.5g,加乙醇30ml,加热回流30分钟,放冷,滤过,滤液蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液。另取槲寄生对照药材1.5g,同法制成对照药材溶液。再取齐墩果酸对照品,加无水乙醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液和对照药材溶液各4μl、对照品溶液2μl,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯-冰醋酸(20:6:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在80℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;再置紫外光灯(365nm)下检视,显相同颜色的荧光斑点。 检查| [font=宋体] 杂质 不得过2%(通则2301)。 水分 不得过12.0%(通则0832第二法)。 总灰分 不得过9.0%(通则2302)。 酸不溶性灰分 不得过2.5%(通则2302)。 【浸出物】 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用乙醇作溶剂,不得少于20.0%。 【含量测定】 照高效液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.1%磷酸溶液(15:85)为流动相;检测波长为264nm。理论板数按紫丁香苷峰计算应不低于5000。 对照品溶液的制备 取紫丁香苷对照品适量,精密称定,加甲醇制成每1ml含50μg的溶液,即得。 供试品溶液的制备 取本品细粉约2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率300W,频率25kHz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。 本品按干燥品计算,含紫丁香苷(C17H2409)不得少于0.040%。 饮片| 【炮制】 除去杂质,略洗,润透,切厚片,干燥。 本品呈不规则的厚片。茎外皮黄绿色、黄棕色或棕褐色。切面皮部黄色,木部浅黄色,有放射状纹理,髓部常偏向一边。叶片黄绿色或黄棕色,全缘,有细皱纹;革质。气微,味微苦,嚼之有黏性。 【含量测定】 同药材,含紫丁香苷(C17H2409)不得少于0.025%。 【鉴别】(除茎横切面外) 【检查】(水分 总灰分) 【浸出物】 同药材。 【性味与归经】 苦,平。归肝、肾经。 【功能与主治】 祛风湿,补肝肾,强筋骨,安胎元。用于风湿痹痛,腰膝酸软,筋骨无力,崩漏经多,妊娠漏血,胎动不安,头晕目眩。 【用法与用量】 9~15g。 【贮藏】 置干燥处,防蛀。
益母草具有祛瘀生新,调经活血,素有“经产良药”之称。益母草主要有效成益母草碱、盐酸水苏碱等,益母草中盐酸水苏碱的含量测定方法较多,主要有分光光度法[1],高效液相色谱法[2,3],薄层扫描法[4]等。《中国药典》2000年版 一部[1]益母草项下对益母草规定了总生物碱的含量,采用的测定方法是分光光度法,但该方法的重现性不好。笔者曾参考文献资料报道[2,3],采用高效液相色谱法试验,文献记载中所用的条件也均未能有较好的重现性,《中国药典》 2005年版 一部[1]益母草项下的含量测定方法采用了双波长薄层色谱扫描法,但其展开剂为乙酸乙酯-正丁醇-盐酸(1:8:3),展开速度极慢,本试验对所用展开剂及显色剂、显色方法加以改进,取得满意结果,现报告如下。 1 仪器、药品与试剂 CS-930型双波长薄层扫描仪,日本岛津公司生产;硅胶G薄层预制板,青岛海洋化工厂分厂生产;盐酸水苏碱对照品(含量测定用,批号:110712-200306),购自中国药品生物制品检定所;所用试剂均为分析纯;益母草(Leonurus japonicus Houtt.)药材购自山西太原万民大药房(经山西中医学院中药鉴定教研室鉴定)。 2 方法与结果 2.1 供试品溶液的制备 取益母草0.5g,精密称定,置具塞三角瓶中,精密加入乙醇50ml,称定重量,超声处理30min,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液25ml,置蒸发皿中,于水浴上蒸干,用盐酸-甲醇(1:100)溶解并转移至5ml量瓶内,加盐酸-甲醇(1:100)至刻度,摇匀,作为供试品溶液。 2.2 对照品溶液的制备 精密称取经105℃干燥至恒重的盐酸水苏碱对照品20mg至20ml量瓶中,加盐酸-甲醇(1:100)溶液使溶解,并稀释至刻度,摇匀,即得(每1ml中含盐酸水苏碱1mg),作为对照品溶液。再分别取1ml,2.5ml,5 ml,7.5ml用盐酸-甲醇(1:100)溶液稀释至10ml,即得到0.1mg/ml,0.25mg/ml,0.5 mg/ml,0.75mg/ml的对照品溶液。 2.3 展开、显色及扫描条件选择 分别吸取盐酸水苏碱对照品溶液及供试品溶液,点于同一硅胶G薄层板上,以丙酮-无水乙醇-盐酸(10:10:1)为展开剂,展开,取出,晾干,于100℃加热10min,取出,放冷,喷以稀碘化铋钾-1%三氯化铁乙醇(2:1)试液,冷风吹至斑点显色清晰。进行光谱扫描,最大吸收波长λs=510mm,参比波长λR=700nm,SX=3。 2.4 线性关系的考察 精密吸取不同浓度(0.1mg/ml,0.25mg/ml,0.5mg/ml,0.75mg/ml,1mg/ml)的对照品溶液各5μl,分别点于同一硅胶G薄层板上,以丙酮-无水乙醇-盐酸(10:10:1)为展开剂,展开,取出,晾干,于100℃加热10min,取出,放冷,喷以稀碘化铋钾-1%三氯化铁乙醇(2:1)试液,冷风吹至斑点显色清晰。照薄层色谱法(《中国药典》 2000年版 一部 附录Ⅵ B)进行扫描, 以点样量为横坐标,吸收度积分值为纵坐标作图,得一直线,回归方程:Y=10869X-1973.9,r=0.9991,盐酸水苏碱点样量在1.0~10.0μg范围内呈良好线性。 2.5 精密度试验 2.5.1 异板精密度试验 精密吸取盐酸水苏碱对照品溶液4μl,分别在不同的硅胶G薄层板上点样,展开,晾干,加热,放冷,显色,扫描,测定吸收度积分值RSD%=2.15。 2.5.2 同板精密度试验 分别精密吸取盐酸水苏碱对照品溶液4μl,在同一块硅胶G薄层板上点5点,展开,取出,晾干,加热,放冷,显色,扫描,测定吸收度积分值RSD%=0.82。 2.6 稳定性试验 取供试品溶液在0、0.5、1.5、2.0h分别点样5μl ,测定样品中盐酸水苏碱斑点吸收峰面积积分值,RSD%为2.61。试验结果表明,经显色后斑点在2h内稳定。 2.7 重复性试验 按拟定的含量测定方法,对同一批样品分别制备5份供试品溶液,精密吸取5μl,点样, 测定斑点吸收峰面积积分值并计算含量。益母草中盐酸水苏碱的含量平均为0.10,RSD=1.90 %。 2.8 回收率试验 采用加样回收法试验。精密称取已知含量的益母草约0.25 g, 精密加入盐酸水苏碱对照品溶液(1mg/ml)1ml,按样品测定项下操作,计算回收率。结果盐酸水苏碱的平均回收率为97.24%, RSD%为1.75%(n=5)。 2.9 样品测定 精密吸取对照品溶液2μl、6μl和供试品溶液10μl,交叉点于同一硅胶G薄层板上,按上述条件测定斑点吸收峰面积的积分值,以外标二点法计算含量,结果益母草中盐酸水苏碱的平均含量为0.50%,RSD为2.39%(n =6)。 3 讨论 《中国药典》2000年版一部[1]益母草项下对益母草规定了总生物碱的含量,采用的测定方法是分光光度法,但无法重复。笔者曾对文献记载的高效液相色谱法所用的条件反复试验,均未能有较好的重现性。彭维[5]明确提出所有报道益母草含量测定中所用的高效液相色谱法,没有方法能够重复出来。本试验采用双波长薄层色谱扫描法,并对展开剂、显色剂及显色条件加以改进,结果满意。可为益母草及其制剂的含量测定提供有效的方法。 《中国药典》 2005年版一部[1]益母草含量测定项下展开剂为乙酸乙酯-正丁醇-盐酸(1:8:3),但展开速度缓慢,本实验以丙酮-无水乙醇-盐酸(10:10:1)为展开剂展开,速度快,分离效果好。 取供试品溶液及对照品溶液点样,展开,取出之后,必需加热至展开剂挥尽再显色,斑点在2h内稳定,否则影响显色效果。 【参考文献】 1 国家药典委员会.中华人民共和国药典.北京:化学工业出版社,2000,一部,237-238;203-204. 2 戚建中. 产妇康颗粒中盐酸水苏碱的HPLC分析.中成药, 2001,23(1):16-18. 3 姜舜尧.益母草药材中水苏碱成分的高效液相色谱法分析.药物分析杂志,2001,(4):21. 4 章曙丹. 薄层扫描法测定鲜母益胶囊中盐酸水苏碱的含量.中成药,2004,26(4):附7. 5 彭维. 产妇康颗粒质量标准研究.中药材,2003,26(3):198-200. (编辑:宋 冰) 作者单位: 030024 山西太原,山西中医学院中药系
蕲蛇 Qishe 本品为蝰科动物五步蛇Agkistrodon acutus (Güenther)的干燥体。多于夏、秋二季捕捉,剖开蛇腹,除去内脏,洗净,用竹片撑开腹部,盘成圆盘状,干燥后拆除竹片。 性状| 本品卷呈圆盘状,盘径17~34cm,体长可达2m。头在中间稍向上,呈三角形而扁平,吻端向上,习称“翘鼻头”。上腭有管状毒牙,中空尖锐。背部两侧各有黑褐色与浅棕色组成的“V”形斑纹17~25个,其“V”形的两上端在背中线上相接,习称“方胜纹”,有的左右不相接,呈交错排列。腹部撑开或不撑开,灰白色,鳞片较大,有黑色类圆形的斑点,习称“连珠斑”;腹内壁黄白色,脊椎骨的棘突较高,呈刀片状上突,前后椎体下突基本同形,多为弯刀状,向后倾斜,尖端明显超过椎体后隆面。尾部骤细,末端有三角形深灰色的角质鳞片1枚。气腥,味微咸。 【浸出物】 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于10.0%。 饮片| 【炮制】 蕲蛇 去头、鳞,切成寸段。 【性状】本品呈段状,长2~4cm,背部呈黑褐色,表皮光滑,有明显的鳞斑,可见不完整的方胜纹。腹部可见白色的肋骨,呈黄白色、淡黄色或黄色。断面中间可见白色菱形的脊椎骨,脊椎骨的棘突较高,棘突两侧可见淡黄色的肉块,棘突呈刀片状上突,前后椎体下突基本同形,多为弯刀状。肉质松散,轻捏易碎。气腥,味微咸。 【鉴别】 聚合酶链式反应法。 模板DNA提取 取本品0.5g,置乳钵中,加液氮适量,充分研磨使成粉末,取0.1g,置1.5ml离心管中,加入消化液275μl,在55℃水浴保温1小时,加入裂解缓冲液250μl,混匀,加到DNA纯化柱中,离心(转速为每分钟10000转)3分钟;弃去过滤液,加入洗脱液800μl,离心(转速为每分钟10000转)1分钟;弃去过滤液,用上述洗脱液反复洗脱3次,每次离心(转速为每分钟10000转)1分钟;弃去过滤液,再离心2分钟,将DNA纯化柱转移入另一离心管中,加入无菌双蒸水100μl,室温放置2分钟后,离心(转速为每分钟10000转)2分钟,取上清液,作为供试品溶液[i],置零下20℃保存备用。另取蕲蛇对照药材0.5g,同法制成对照药材模板DNA溶液。 PCR反应鉴别引物:5'GGCAATTCACTACACAGCCAA-CArCAACT3'和5' CCATAGTCAGGTGGTTAGTGATAC 3'。PCR反应体系:在200μl离心管中进行,反应总体积为25μl,反应体系包括10×PCR缓冲液 2.5μl,dNTP(2.5mmol/L)2μl,鉴别引物(10μmol/L)各0.5μl,高保真Taq DNA聚合酶(5U/μl)0.2μl,模板0.5μl,无菌双蒸水18.8μl。将离心管置PCR仪,PCR反应参数:95℃预变性5分钟,循环反应30次(95℃30秒,63℃45 秒),延伸(72℃)5分钟。 电泳检测 照琼脂糖凝胶电泳法方法2(通则0541),胶浓度为1%,胶中加入核酸凝胶染色剂GelRed;供试品与对照药材PCR反应溶液的上样量分别为8μl,DNA分子量标记上样量为2μl(0.5μg/μl)。电泳结束后,取凝胶片在凝胶成像仪上或紫外透射仪上检视。供试品凝胶电泳图谱中,在与对照药材凝胶电泳图谱相应的位置上,在300~400bp应有单一DNA条带。 【检查】 水分 不得过14.0%(通则0832第二法)。 【浸出物】 同药材,不得少于12.0% 蕲蛇肉 去头,用黄酒润透后,除去鳞、骨,干燥。 【性状】本品呈条状或块状,长2~5cm,可见深黄色的肉条及黑褐色的皮。肉条质地较硬,皮块质地较脆。有酒香气,味微咸。 【鉴别】 同蕲蛇(饮片)。 【检查】 水分 不得过14.0%(通则0832第二法)。 总灰分 不得过4.0%(通则2302) 【浸出物】 同药材,不得少于12.0%。 酒蕲蛇 取净蕲蛇段,照酒炙法(通则0213)炒干。 每100kg蕲蛇,用黄酒20kg。 【性状】 本品形如蕲蛇段,表面棕褐色或黑色,略有酒气。气腥,味微咸。【鉴别】 同蕲蛇(饮片)。 【检查】 水分 不得过14.0%(通则0832第二法)。 【浸出物】 同药材,不得少于12.0% 【性味与归经】 甘、咸,温;有毒。归肝经。 【功能与主治】 祛风,通络,止痉。用于风湿顽痹,麻木拘挛,中风口眼斜,半身不遂,抽搐痉挛,破伤风,麻风,疥癣。 【用法与用量】 3~9g;研末吞服,一次1~1.5g,一日2~3次。 【贮藏】 置干燥处,防霉,防蛀。
蒺藜 Jili 本品为蒺藜科植物蒺藜Tribulus terrestris L.的干燥成熟果实。秋季果实成熟时采割植株,晒干,打下果实,除去杂质。 性状| 本品由5个分果瓣组成,呈放射状排列,直径7~12mm。常裂为单一的分果瓣,分果瓣呈斧状,长3~6mm;背部黄绿色,隆起,有纵棱和多数小刺,并有对称的长刺和短刺各1对,两侧面粗糙,有网纹,灰白色。质坚硬。气微,味苦、辛。 鉴别| (1)本品粉末黄绿色。内果皮纤维木化,上下层纵横交错排列,少数单个散在,有时纤维束与石细胞群相连结。中果皮纤维多成束,多碎断,直径15~40μm,壁甚厚,胞腔疏具圆形点状纹孔。石细胞长椭圆形或类圆形,黄色,成群。种皮细胞多角形或类方形,直径约30μm,壁网状增厚,木化。草酸钙方晶直径8~20μm。 (2)取本品粉末3g,加三氯甲烷50ml,超声处理30分钟,滤过,弃去三氯甲烷液,药渣挥干,加水1ml,搅匀,加水饱和的正丁醇50ml,超声处理30分钟,分取上清液,加2倍量的氨试液洗涤,弃去洗液,取正丁醇液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取蒺藜对照药材3g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13:7:2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以改良对二甲氨基苯甲醛溶液(取对二甲氨基 苯甲醛1g,加盐酸34ml,甲醇100ml,摇匀,即得),在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。 检查|水分 不得过9.0%(通则0832第二法)。 总灰分 不得过12.0%(通则2302)。 【含量测定】 对照品溶液的制备 取蒺藜苷元对照品适量,精密称定,加甲醇制成每1ml含0.15mg的溶液,既得。 标准曲线的制备 标准曲线的制备精密量取对照品溶0.1ml、0.2ml、0.3m1、0.4ml、0.5m1、0.6ml,分别置具塞试管中,置水浴中挥干溶剂,精密加入高氯酸5ml,摇匀,置60℃水浴保温15分钟,取出后立即冰水浴冷却至室温,以相应的试剂为空白,照紫外-可见分光光度法(通则0401),在285nm波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。 测定法 取本品细粉0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,加热回流2小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密吸取续滤液10ml,回收溶剂至干,残渣加正丁醇饱和的水10ml解,用水饱和正丁醇振摇提取5次,每次10ml,合并正丁醇液,用氨试液洗涤2次,每次5ml,弃去氨试液,正丁醇液回收溶剂至干。残渣加80%甲醇溶解,转移至50ml量瓶中,加80%甲醇至刻度,摇匀。精密量取1~2ml,置10ml具塞试管中,照标准曲线的制备项下的方法,自“置水浴中挥干溶剂”起,同法操作,依法测定吸光度,从标准曲线上读出供试品溶液中相当于蒺藜苷元的重量,计算,即得。 本品按干燥品计算,含蒺藜总皂苷以蒺藜苷元(C27H38O4)计,不得少于1.0%。 饮片| 【炮制】 蒺藜 除去杂质。 【性状】 【鉴别】 【检查】【含量测定】 同药材。 炒蒺藜 取净蒺藜,照清炒法(通则0213)炒至微黄色。 本品多为单一的分果瓣,分果瓣呈斧状,长3~6mm;背部棕黄色,隆起,有纵棱,两侧面粗糙,有网纹。气微香,味苦、辛。 【鉴别】 【检查】 同药材。 【性味与归经】 辛、苦,微温;有小毒。归肝经。 【功能与主治】 平肝解郁,活血祛风,明目,止痒。用于头痛眩晕,胸胁胀痛,乳闭乳痈,目赤翳障,风疹瘙痒。 【用法与用量】 6~10g。 【贮藏】 置干燥处,防霉。
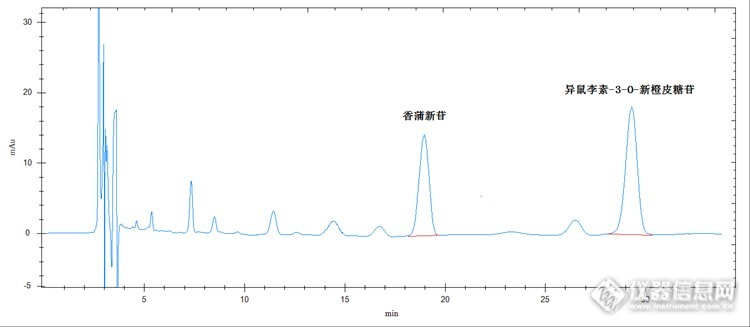
好药材不怕检,蒲黄药材接着检 蒲黄药材为香蒲科植物狭叶香蒲、宽叶香蒲、东方香蒲和长苞香的花粉,具有止血,化瘀,通淋功效。用于吐血,衄血,咯血,崩漏,外伤出血,经闭痛经,脘腹刺痛,跌扑肿痛,血淋涩痛效果较好。实验部分原理 取适量该药材,加甲醇溶解,加热回流或超声波提取,经进样系统进样,色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量计算。仪器及试剂 仪器:高效液相色谱仪(紫外检测器),柱温箱,超声波清洗仪,溶剂过滤器,针筒式过滤器,加热回流装置,电子天平 试剂:甲醇(色谱纯),超纯水样品制备 对照品溶液的制备:精密称取异鼠李素-3-O-新橙皮糖苷、香蒲新苷对照品适量,加甲醇配制成浓度均为50μg/ml的对照品溶液,备用。 供试品溶液的制备:精密称取本品约0.5g,置具塞锥形瓶中,精密加入50ml甲醇后,称定重量并记录,冷浸12小时后加热回流1小时(或超声波超声30min),放冷,再次称定重量,用甲醇补足减少的重量,摇匀,滤过,待测。色谱条件检测器:紫外检测器色谱柱:C18,4.6 X 250mm,5μm流动相:乙腈:0.05%磷酸溶液=15:85(V:V)检测波长:254nm进样量20μl柱温:室温对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192152_519002_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519003_2498430_3.png 从以上色谱图我们可以看出样品出峰时间很晚,峰形也较差。下面我们换用一根耐酸性色谱柱,效果我们请看色谱图。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519004_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519005_2498430_3.png 换了这根色谱柱,色谱图的峰形好了很多,出峰时间也明显有所提前,但保留时间还是有点晚。下面我们又把色谱柱温度调整了一下,调到了40℃,效果接着往下看。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519006_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410231859_519702_2498430_3.png 当然保留时间还可以再缩短缩短(通过增加流动相中甲醇含量,或提高高压泵流速,或换用更高效更短的色谱柱),但供试品中异鼠李素-3-O-新橙皮糖苷的附近有一个干扰物,为了保证分离度,这个分析时间已经比较合理,不要再缩短了。 检测蒲黄药材的这个方法到现在已经很完美了。但有几点事项需要注意。1.样品若采用超声波超声提取,为了保证提取效果有时得增加超声时间或超声波水域温度。2.检测这个样品最好要选择一款效果好的色谱柱,如耐酸性的色谱柱。3.为了缩短检测时间我们可以升高色谱柱的温度,对于这个样品效果就挺好。当然适当增加流动相中甲醇含量或增加高压泵流速,或换用更高效更短的色谱柱也能达到比较理想的效果。
蓖麻子 Bimazi 本品为大戟科植物蓖麻Ricinus communis L.的干燥成熟种子。秋季采摘成熟果实,晒干,除去果壳,收集种子。 性状| 本品呈椭圆形或卵形,稍扁,长0.9~1.8cm,宽0.5~1cm。表面光滑,有灰白色与黑褐色或黄棕色与红棕色相间的花斑纹。一面较平,一面较隆起,较平的一面有1条隆起的种脊;一端有灰白色或浅棕色突起的种阜。种皮薄而脆。胚乳肥厚,白色,富油性,子叶2,菲薄。气微,味微苦辛。 鉴别| (1)本品粉末灰黄色或黄棕色。种皮栅状细胞红棕色,细长柱形,排列紧密,孔沟细密,胞腔内含红棕色物质。外胚乳组织细胞壁不明显,密布细小圆簇状结晶体,菊花形或圆球形,直径8~20mm。内胚乳细胞类多角形,胞腔内含糊粉粒和脂肪油滴。 (2)取本品粗粉1g,加无水乙醇10ml,冷浸30分钟,滤过,取滤液作为供试品溶液。另取蓖麻子对照药材1g,同法制成对照药材溶液。再取蓖麻酸对照品,加无水乙醇制成每1ml含1μl的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液和对照药材溶液各1μl、对照品溶液2μl,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲酸(14:4:0.4)为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,在110℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。 检查| 水分 不得过7.0%(通则0832第二法)。 酸败度 照酸败度测定法(通则2303)测定。 酸值 不得过35.0。 羰基值 不得过7.0。 过氧化值 不得过0.20。 蓖麻碱 照高效[url=https://insevent.instrument.com.cn/t/5p]液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-水-二乙胺(11:89:0.03)为流动相;检测波长为307nm。理论板数按蓖麻碱峰计算应不低于3000。 对照品溶液的制备 取蓖麻碱对照品适量,精密称定,加甲醇制成每1ml含0.125mg的溶液,即得。 供试品溶液的制备 取本品粉末(过二号筛)约2.5g,精密称定,置索氏提取器中,加石油醚(60~90℃)适量,加热回流提取4小时,弃去石油醚液,药渣挥去溶剂,转移至具塞锥形瓶中,精密加入50%甲醇50ml,称定重量,加热回流2小时,放冷,称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。 本品按干燥品计算,含蓖麻碱(C8H8N2O2)不得过0.32%。 饮片| 【炮制】 用时去壳,捣碎。 【性状】 【鉴别】 【检查】 同药材。 【性味与归经】 甘、辛,平;有毒。归大肠、肺经。 【功能与主治】 泻下通滞,消肿拔毒。用于大便燥结,痈疽肿毒,喉痹,瘰疬。 【用法与用量】 2~5g。外用适量。 【贮藏】 置阴凉干燥处。
蟾酥 Chansu 本品为蟾蜍科动物中华大蟾蜍Bufo bufo gargarizans Cantor 或黑眶蟾蜍Bufo melanostictus Schneider 的干燥分泌物。多于夏、秋二季捕捉蟾蜍,洗净,挤取耳后腺和皮肤腺的白色浆液,加工,干燥。 性状| 本品呈扁圆形团块状或片状。棕褐色或红棕色。团块状者质坚,不易折断,断面棕褐色,角质状,微有光泽;片状者质脆,易碎,断面红棕色,半透明。气微腥,味初甜而后有持久的麻辣感,粉末嗅之作嚏。 鉴别| (1)本品断面沾水,即呈乳白色隆起。 (2)取本品粉末0.1g,加甲醇5ml,浸泡1小时,滤过,滤液加对二甲氨基苯甲醛固体少量,滴加硫酸数滴,即显蓝紫色。 (3)取本品粉末0.1g,加三氯甲烷5ml,浸泡1小时,滤过,滤液蒸干,残渣加醋酐少量使溶解,滴加硫酸,初显蓝紫色,渐变为蓝绿色。 (4)取〔含量测定〕项下供试品溶液10ml,水浴蒸干,用甲醇2ml溶解,作为供试品溶液。另取蟾酥对照药材0.2g,加甲醇10ml,加热回流30分钟,滤过,滤液作为对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各10l,分别点于同一硅胶[/font]G薄层板上,以环己烷-三氯甲烷-丙酮4:3:3)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点。 【特征图谱】 照高效液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 同〔含量测定〕项。 参照物溶液的制备 取蟾酥对照药材25mg,按〔含量测定〕项下供试品溶液制备方法制成对照药材参照物溶液 另取〔含量测定〕项下的对照品溶液,作为对照品参照物溶液。 供试品溶液的制备 取〔含量测定〕项下的供试品溶液,即得。 测定法 分别精密吸取参照物溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。 供试品特征图谱中应呈现5个特征峰,并应与对照药材参照物色谱峰中的5个特征峰相对应,其中峰4应与华蟾酥毒基参照物峰的保留时间相一致。 检查| 水分 不得过13.0%(通则0832第二法)。 总灰分 不得过5.0%(通则2302[font=宋体])。 酸不溶性灰分 不得过2.0%(通则2302)。 【含量测定】 照高效液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂 以乙腈为流动相A,0.3%乙酸溶液为流动相B,按下表中的规定进行梯度洗脱 柱温为30℃ 流速为每分钟0.6ml 检测波长为296nm。理论板数按华蟾酥毒基峰计算应不低于10000 对照品溶液的制备 取华蟾酥毒基对照品适量,精密称定,加甲醇制成每lml含100μg的溶液,即得。 供试品溶液的制备 取本品细粉约25mg,精密称定,置具塞锥形瓶中,精密加入甲醇20ml,称定重量,加热回流1小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取上述对照品溶液10μl与供试品溶液10~20μl,注入液相色谱仪,测定,以华蟾酥毒基对照品为参照,以其相应的峰为S峰,计算蟾毒灵和脂蟾毒配基的相对保留时间,其相对保留时间应在规定值的±5%范围之内相对保留时间及校正因子见下表: 以华蟾酥毒基对照品为对照,分别乘以校正因子,计算华蟾酥毒基、蟾毒灵和脂蟾毒配基的含量。 本品按干燥品计算,含蟾毒灵(C24H34O4)、华蟾酥毒基(C26H34O6)和脂蟾毒配基(C24H32O4)的总量不得少于7.0%。 饮片| 【炮制】 蟾酥粉 取蟾酥,捣碎,加白酒浸渍,时常搅动至呈稠膏状,干燥,粉碎。 每10kg蟾酥,用白酒20kg。 【性状】 本品为棕黄色至棕褐色粉末。气微腥,味初甜而后有持久的麻辣感,嗅之作嚏。 【检査】 水分 同药材,不得过8.0%。 【鉴别】(2)(3)(4) 【特征图谱】【含量测定】同药材。 【性味与归经】 辛,温;有毒。归心经。 【功能与主治】 解毒,止痛,开窍醒神。用于痈疽疔疮,咽喉肿痛,中暑神昏,痧胀腹痛吐泻。 【用法与用量】 0.015~0.03g,多入丸散用。外用适量。 【注意】 孕妇慎用。 【贮藏】 置干燥处,防潮。
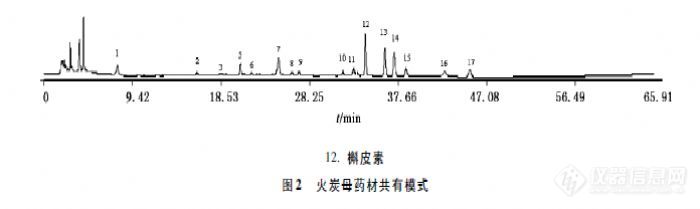
【作者】 王金香;【Author】 WANG Jin-xiang(Guangdong Food and Drug Vocational College,Guangzhou 510520,China)【机构】 广东食品药品职业学院;【摘要】 目的:建立火炭母药材的HPLC指纹图谱分析方法,为火炭母药材质量评价提供参考依据。方法:色谱柱PlatisilODS C18(4.6 mm×250 mn,5μm),流动相乙腈-0.05%磷酸溶液,梯度洗脱进行色谱分离;洗脱时间60 min,流速1.0 mL.min-1,检测波长360 nm。结果:确定17个共有色谱峰为特征峰构成火炭母药材HPLC指纹图谱,相似度评价结果表明,各产地火炭母药材相似度均在0.90以上。结论:所用方法准确可靠,所得指纹图谱共有模式可作为火炭母药材质量控制的依据。http://ng1.17img.cn/bbsfiles/images/2012/07/201207181207_378450_1761902_3.jpg
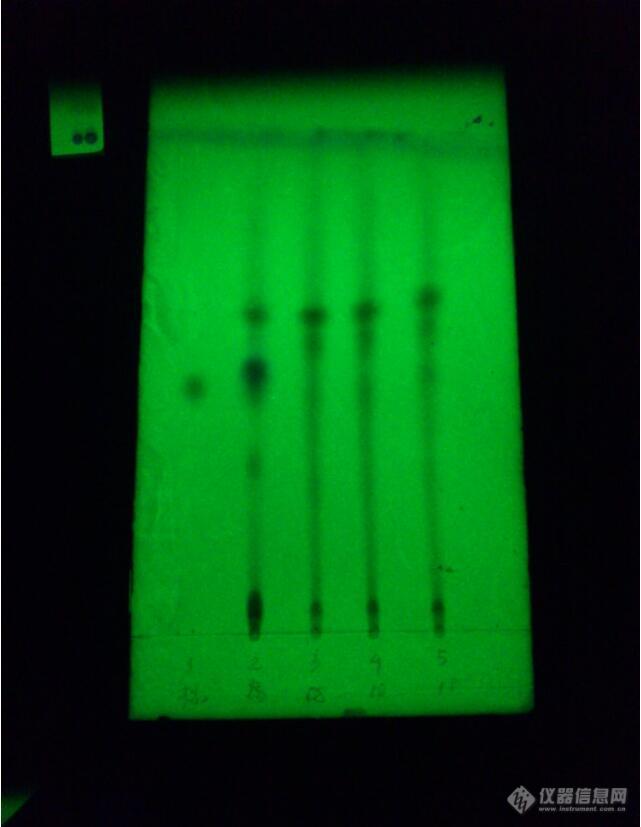
按照药典规定的,展开剂:氯仿-醋酸乙酯-丙酮-甲酸=6:2.5:2.5:0.21为原儿茶酸对照品2药材对照品3、4、5均为制成的中药制剂供试品。我已经增大浓度试过了,色谱图无差异[img=,641,827]https://ng1.17img.cn/bbsfiles/images/2019/08/201908140947125804_8826_1791505_3.jpg!w641x827.jpg[/img]
作者: 张启伟,张永欣,孙玉茹,张 东(中国中医研究院,中药研究所,北京,100700)摘要: 目的: 建立路路通药材中桦木酮酸的鉴别及含量测定方法方法: 薄层色谱法鉴别药材用醋酸乙酯提取在以羧甲基纤维素钠为粘合剂的硅胶G 薄层板上用石油醚(60-90 ) 丙酮(17 3)展开,磷钼酸显色高效液相色谱法进行含量测定样品用无水乙醇超声提取Diamonsil C18 柱, 甲醇-水-冰乙酸 (87:13:0.1)为流动相, 流速1.0ml min-1 ,柱温35 ,蒸发光散射检测器蒸发管温度82 气体流速1.25L min-1 结果: 薄层层析鉴别重现性好在上述液相色谱条件下桦木酮酸获较好分离, 进样量在0.65- 3.25 g 范围其对数与峰面积对数呈线性关系( r=0.9997), 加样回收率为98.0%, 重现性为2.5% 对收集的11 批商品药材进行了定性鉴别和含量测定结论: 本研究为路路通质量标准的建立提供可借鉴的方法.谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208141931_383831_1609970_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208141931_383832_1609970_3.jpg
鉴别| (1)本品粉末黄绿色。上表皮细胞垂周壁呈波状弯曲,气孔不定式。下表皮细胞垂周壁平直,无气孔。通气组织多破碎,由薄壁细胞组成,细胞间隙较大。草酸钙簇晶较小。草酸钙针晶成束。 (2)取本品粉末1g,加甲醇10ml,超声处理30分钟,放置,取上清液作为供试品溶液。另取浮萍对照药材1g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各2μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-丁酮-甲酸-水(6:3:1:1)为展开剂,展开,取出,晾干,喷以1%三氯化铝无水乙醇溶液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。 检查| 水分 不得过8.0%(通则0832第二法)。 图片 其他| 【性味与归经】 辛,寒。归肺经。 【功能与主治】 宣散风热,透疹,利尿。用于麻疹不透,风疹瘙痒,水肿尿少。 【用法与用量】 3~9g。外用适量,煎汤浸洗。 【贮藏】 置通风干燥处,防潮。

准确测定芦荟药材中芦荟苷含量 芦荟大家都知道,是一种植物,常被当做观赏品花盆养殖,属于为百合科植物。芦荟药材是由芦荟叶的汁液浓缩成的干燥物。 大家知道芦荟长的有特点,很漂亮,殊不知它还有很多药物功能。芦荟药材气味较特殊,有特殊臭气,味极苦。具有泻下通便、清肝泻火、杀虫疗疳等药物功效。可用于治疗热结便秘、惊痫抽搐、小儿疳积、外治癣疮、蚊虫叮咬等病症。另外它切成薄片贴在脸上或身上,还可以保湿、美容,还可以炒着吃,清凉、降火等。实验部分【原理】 精密称取适量芦荟样品经甲醇溶解,超声提取后注入高效液相色谱系统,C18色谱柱分离,紫外检测器检测,外标法(保留时间定性,峰面积定量)计算,得出该样品中芦荟苷含量。【仪器及试剂】 仪器:高效液相色谱仪(紫外检测器+等度泵+柱温箱+在线脱气机等),超声波清洗仪,溶剂过滤器,电子天平(0.0001),药典筛(五号筛)等。 试剂:甲醇(色谱纯),乙腈(色谱纯),超纯水等。【样品制备】 对照品溶液制备:准确称取芦荟苷对照品2.5mg于25ml容量瓶中,加甲醇溶解、定容,制成0.1mg/ml芦荟苷对照品溶液,备用。 供试品溶液制备:取芦荟样品适量,充分粉碎后过药典筛五号筛,准确称取过筛粉末0.1g于100ml容量瓶中,加甲醇适量,超声处理30分钟,放冷,加甲醇至刻度,摇匀,0.45um微膜滤过,待测。【色谱条件】检测器:紫外检测器检测波长:355nm色谱柱:Promosil C18 ( 250 mm X 4.6mm,5μm )流动相:乙腈:水=25:75(V:V)流速:1.0ml/min柱温:30℃进样量:10μl【色谱图】对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/09/201509022207_564407_2536753_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/09/201509022207_564408_2536753_3.png 色谱图效果还不错吧,色谱峰形,分离度都还可以,色谱柱理论塔板数也很高。只可惜按外标法计算,http://ng1.17img.cn/bbsfiles/images/2015/09/201509022207_564409_2536753_3.png该样品中芦荟苷含量只有11%左右,远低于药典要求的18%,不符合药典要求,属于不合格品。【结论】 该高效液相色谱法检测芦荟药材中芦荟苷含量方便、准确,该方法适合该项目检测。
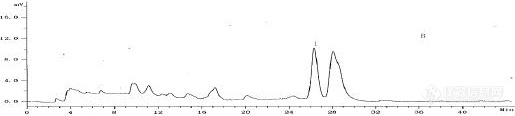
天麻对照品天麻素和对羟基苯甲醇保留时间分别是11和22分钟,天麻药材出峰时间是15和26分钟,且时间还比较稳定,后面更换了新柱子,把柱温箱温度从30度调到35度,把泵从B泵更换到A泵,重新配流动相,供试品,发现还是一样漂移。而且之前做的特征图谱保留时间又对的上。流动相过滤了也超声了。真的不知道该咋办了??
鉴别| (1)粉末灰褐色。石细胞淡黄色或黄绿色,类圆形、类方形、圆多角形或长条形,直径20~50μm,孔沟明显,有的胞腔含暗棕色物。草酸钙砂晶多存在于薄壁细胞中。木纤维多成束,直径12~25μm,具斜纹孔或相交成十字形、人字形。皮层纤维细长,直径12~28μm,微木化,纹孔稀少,有的可见分隔。具缘纹孔导管直径15~90μm,纹孔排列紧密,有的横向延长成梯状,排列整齐。 (2)取本品粉末2g,加甲醇30ml,超声处理30分钟,滤过,滤液蒸干,残渣加无水乙醇2ml使溶解,加入硅胶G 3g,混匀,置水浴上挥干溶剂,加于硅胶G柱(15g,内径为1.5~2cm)上,用环己烷-乙酸乙酯(1:1)混合溶液100ml洗脱,收集洗脱液,蒸干,残渣加乙醇2ml使溶解,作为供试品溶液。另取海风藤对照药材2g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以三氯甲烷-丙酮-甲醇(7:1:0.5)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。 检查| 水分 不得过12.0%(通则0832第二法)。 总灰分 不得过10.0%(通则2302)。 酸不溶性灰分 不得过2.0%(通则2302)。 【浸出物】 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用稀乙醇作溶剂,不得少于10.0%。
蒲公英 Pugongying 本品为菊科植物蒲公英Taraxacum mongolicum Hand.-Mazz.、碱地蒲公英 Taraxacum borealisinense Kitam.或同属数种植物的干燥全草。春至秋季花初开时采挖,除去杂质,洗净,晒干。 性状| 本品呈皱缩卷曲的团块。根呈圆锥状,多弯曲,长3~7cm;表面棕褐色,抽皱;根头部有棕褐色或黄白色的茸毛,有的已脱落。叶基生,多皱缩破碎,完整叶片呈倒披针形,绿褐色或暗灰绿色,先端尖或钝,边缘浅裂或羽状分裂,基部渐狭,下延呈柄状,下表面主脉明显。花茎1至数条,每条顶生头状花序,总苞片多层,内面一层较长,花冠黄褐色或淡黄白色。有的可见多数具白色冠毛的长椭圆形瘦果。气微,味微苦。 鉴别| (1)本品叶表面观:上下表皮细胞垂周壁波状弯曲,表面角质纹理明显或稀疏可见。上下表皮均有非腺毛,3~9细胞,直径17~34μm,顶端细胞甚长,皱缩呈鞭状或脱落。下表皮气孔较多,不定式或不等式,副卫细胞3~6个,叶肉细胞含细小草酸钙结晶。叶脉旁可见乳汁管。 根横切面:木栓细胞数列,棕色。韧皮部宽广,乳管群断续排列成数轮。形成层成环。木质部较小,射线不明显;导管较大,散列。 (2)取本品粉末1g,加80%甲醇10ml,超声处理20分钟,滤过,取滤液作为供试品溶液。另取蒲公英对照药材1g,同法制成对照药材溶液。再取菊苣酸对照品,加80%甲醇制成每1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各4ul、对照品溶液3ul,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲酸-水(6:12:5:2)为展开剂,展开,取出,晾干,喷以1%三氯化铝乙醇溶液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。 检查|水分 不得过13.0%(通则0832第二法)。 【含量测定】 照高效液相色谱法(通则0512)测定。 色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂 以甲醇为流动相A,以0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱 检测波长为327nm。理论板数按菊苣酸峰计算应不低于5000。 对照品溶液的制备 取菊苣酸对照品适量,精密称定,加80%甲醇制成每1ml含0.2mg的溶液,即得。 供试品溶液的制备 取本品粉末(过四号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20ml,称定重量,超声处理(功率400W,频率40kHz)20分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液即得。 测定法 分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,即得。 本品按干燥品计算,含菊苣酸(C22H18O12)不得少于0.45%。 饮片| 【炮制】 除去杂质,洗净,切段,干燥。 【性状】本品为不规则的段。根表面棕褐色,抽皱;根头部有棕褐色或黄白色的茸毛,有的已脱落。叶多皱缩破碎,绿褐色或暗灰绿色,完整者展平后呈倒披针形,先端尖或钝,边缘浅裂或羽状分裂,基部渐狭,下延呈柄状。头状花序,总苞片多层,花冠黄褐色或淡黄白色。有时可见具白色冠毛的长橢圆形瘦果。气微,味微苦。 【检查】 水分 同药材,不得过10.0%。 【浸出物】 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用75%乙醇作溶剂,不得少于18.0%。 【含量测定】 同药材,含菊苣酸(C22H18O12)不得少于0.30%。。 【鉴别】 同药材。 【性味与归经】 苦、甘,寒。归肝、胃经。 【功能与主治】 清热解毒,消肿散结,利尿通淋。用于疔疮肿毒,乳痈,瘰疬,目赤,咽痛,肺痈,肠痈,湿热黄疸,热淋涩痛。 【用法与用量】 10~15g。 【贮藏】 置通风干燥处,防潮,防蛀。
中药材鉴别之紫苏子 紫苏子 Zisuzi https://ng1.17img.cn/bbsfiles/images/2024/10/202410241019516963_3736_6561489_3.png!w690x690.jpg 本品为唇形科植物紫苏Perilla frutescens(L.)Britt.的干燥成熟果实。秋季果实成熟时采收,除去杂质,晒干。 性状| 本品呈卵圆形或类球形,直径约1.5mm。表面灰棕色或灰褐色,有微隆起的暗紫色网纹,基部稍尖,有灰白色点状果梗痕。果皮薄而脆,易压碎。种子黄白色,种皮膜质,子叶2,类白色,有油性。压碎有香气,味微辛。 鉴别| (1)本品粉末灰棕色。种皮表皮细胞断面观细胞极扁平,具钩状增厚壁;表面观呈类椭圆形,壁具致密雕花钩纹状增厚。外果皮细胞黄棕色,断面观细胞扁平,外壁呈乳突状;表面观呈类圆形,壁稍弯曲,表面具角质细纹理。内果皮组织断面观主为异型石细胞,呈不规则形;顶面观呈类多角形,细胞间界限不分明,胞腔星状。内胚乳细胞大小不一,含脂肪油滴;有的含细小草酸钙方晶。子叶细胞呈类长方形,充满脂肪油滴。 (2)取本品粉末1g,加甲醇25ml,超声处理30分钟,滤过,滤液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取紫苏子对照药材1g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各2μl,分别点于同一硅胶G薄层板上,以正己烷-甲苯-乙酸乙酯-甲酸(2:5:2.5:0.5)为展开剂,展开,取出,晾干,喷以三氯化铝试液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。 饮片| 水分 不得过8.0%(通则0832第二法)。 【含量测定】 照高效[url=https://insevent.instrument.com.cn/t/5p]液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.1%甲酸溶液(40:60)为流动相;检测波长为330nm。理论板数按迷迭香酸峰计算应不低于3000。 对照品溶液的制备 取迷迭香酸对照品适量,精密称定,加甲醇制成每1ml含80μg的溶液,即得。 供试品溶液的制备 取本品粉末(过二号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,密塞,称定重量,加热回流2小时,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液10μl与供试品溶液20μl,注入液相色谱仪,测定,即得。 本品按干燥品计算,含迷迭香酸(C18H16O8)不得少于0.25%。 饮片| 【炮制】 紫苏子 除去杂质,洗净,干燥。 【性状】 【鉴别】 【检查】 【含量测定】 同药材。 炒紫苏子 取净紫苏子,照清炒法(通则0213)炒至有爆声。 本品形如紫苏子,表面灰褐色,有细裂口,有焦香气。 【检查】 水分 同药材,不得过2.0%。 【含量测定】 同药材,含迷迭香酸(C18H16O8)不得少于0.20%。 【鉴别】 同药材。 【性味与归经】 辛,温。归肺经。 【功能与主治】 降气化痰,止咳平喘,润肠通便。用于痰壅气逆,咳嗽气喘,肠燥便秘。[/font] 【用法与用量】 3~10g。 【贮藏】 置通风干燥处,防蛀。
蔓荆子 Manjingzi https://ng1.17img.cn/bbsfiles/images/2024/10/202410241037097660_5040_6561489_3.png!w690x690.jpg 本品为马鞭草科植物单叶蔓荆Vitex trifolia L.Var.simplicifolia Cham.或蔓荆Vitex trifolia L.的干燥成熟果实。秋季果实成熟时采收,除去杂质,晒干。 性状| 本品呈球形,直径4~6mm。表面灰黑色或黑褐色,被灰白色粉霜状茸毛,有纵向浅沟4条,顶端微凹,基部有灰白色宿萼及短果梗。萼长为果实的1/3~2/3,5齿裂,其中2裂较深,密被茸毛。体轻,质坚韧,不易破碎,横切面可见4室,每室有种子1枚。气特异而芳香,味淡、微辛。 鉴别| (1)本品粉末灰褐色。花萼表皮细胞类圆形,壁多弯曲;非腺毛2~3细胞,顶端细胞基部稍粗,有疣突。外果皮细胞多角形,有角质纹理和毛茸脱落后的痕迹,并有腺毛与非腺毛:腺毛分头部单细胞、柄1~2细胞及头部2~6细胞、柄单细胞两种;非腺毛2~4细胞,长14~68μm,多弯曲,有壁疣。中果皮细胞长圆形或类圆形,壁微木化,纹孔明显。油管多破碎,含分泌物,周围细胞有淡黄色油滴。内果皮石细胞椭圆形或近方形,直径10~35μm。种皮细胞圆形或类圆形,直径42~73μm,壁有网状纹理,木化。 (2)取本品粉末5g,加石油醚(60~90℃)50ml,加热回流2小时,滤过,弃去石油醚液,药渣挥干,加丙酮80ml,加热回流1.5小时,滤过,滤液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。另取蔓荆子黄素对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5μl,分别点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,以环己烷-乙酸乙酯-甲醇(3:2:0.2)为展开剂,展开,取出,晾干,喷以10%三氯化铝乙醇溶液。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。 [font=宋体] 检查| 杂质 不得过2%(通则2301)。 水分 不得过14.0%(通则0832第四法)。 总灰分 不得过 7.0%(通则 2302)。 【浸出物】 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用甲醇作溶剂,不得少于8.0% 。 【含量测定】 照高效液相色谱法(通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.4%磷酸溶液(60:40)为流动相;检测波长为258nm。理论板数按蔓荆子黄素峰计算应不低于2000。 对照品溶液的制备 取蔓荆子黄素对照品适量,精密称定,加甲醇制成每1ml含30μg的溶液,即得。 供试品溶液的制备 取本品粉末(过三号筛)约2g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,加热回流1小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。 本品按干燥品计算,含蔓荆子黄素(C19H18O8)不得少于0.030%。 饮片| 【炮制】 蔓荆子 除去杂质。 【性状】 【鉴别】 【检查】(水分 总灰分) 【浸出物】【含量测定】 同药材。 炒蔓荆子 取净蔓荆子,照清炒法(通则0213)微炒。用时捣碎。 本品形如蔓荆子,表面黑色或黑褐色,基部有的可见残留宿萼和短果梗。气特异而芳香,味淡、微辛。 【检查】 水分 同药材,不得过7.0%。 【鉴别】(2) 【检查】(总灰分) 【浸出物】 【含量测定】 同药材。 【性味与归经】 辛、苦,微寒。归膀胱、肝、胃经。 【功能与主治】 疏散风热,清利头目。用于风热感冒头痛,齿龈肿痛,目赤多泪,目暗不明,头晕目眩。 【用法与用量】 5~10g。 【贮藏】 置阴凉干燥处。
问题: 麻烦各位大师,药典上要求一个药材两个波长,检测两个对照品混合液。计算的时候应该怎么计算?回复: 分别各个波长的处理图谱,分别算
问题: 麻烦各位大师,药典上要求一个药材两个波长,检测两个对照品混合液。计算的时候应该怎么计算?回复: 分别各个波长的处理图谱,分别算
安徽亳州炒药农民三个月净赚超过一年收入2010年11月17日 01:58 第一财经日报 陆晋源 上午8点10分,亳州中药材交易市场铁门一开,阵阵人流涌进两个楼层,昨夜锁上的药箱和麻袋被陆续打开,原本空旷的市场瞬间人声鼎沸。 位于皖西北的安徽亳州,有“药都”之称,全城60%以上人口从事中药材生意,中国最大的药材交易市场就坐落于此。 中国中药协会上周发布的一份报告显示,过去一年里,中药中采用的草本植物,逾四分之一的价格上涨了50%至100%,少数药草的价格涨幅在300%以上。 中药材的价格走势,历来就是亳州地面上最热门的话题,但今年,人们更渴望一夜暴富。 当地一名公务员告诉《第一财经日报》记者,他表兄从事中药材生意,年初买下了200多吨的天麻,“26块钱买进,180块卖出,整整赚了几百万啊!”转念想到自己每月不满3000元的工资,他不禁叹了口气。 3个月挣够一年收成 在阚治东的回忆录《荣辱二十年》中,上海证券交易所当年的热闹景象,如今仍可在亳州中药材市场上寻到其影子。市场里,男女老少,黑压压的一片,当日价格表、出售和求购的广告牌到处都是,亳州人津津有味地炒着他们自己的“股票”。 奇怪的是,这里很少有交易达成,多数“买家”只是问个价,而摊主似乎也并不急于推销货物。原来,偌大的市场内陈列的只是样品,若双方有意,取货交割都在亳州各地的仓库和冷库中进行,“场外交易,场内询价”,这种颇有些神秘色彩的交易方式在亳州司空见惯。 更奇怪的是,卖药材的是亳州人,询价的也多是亳州人。上述公务员告诉记者,外地来收购药材的老板绝大多数通过中间商,中间商就是亳州本地人,通过替老板砍价进货,赚取微薄的差价和服务费。“老板走几百吨货,他(中间商)从中赚个一万来块钱。” 农民李运领在当地也是一个小炒家。四个月前,李运领还是个老老实实耕田的农民,年收入不到2万,而在今年短短3个月中,他已赚够了以前一年的钱。8月份李运领以20元/公斤买进1吨多的槐米,9月份以30元/公斤卖出,并再次买入1吨多,至10月又以40元/公斤转手,如此净赚近3万元。 “拿我们当地话说,要发发得快,要砍砍得快。”李运领告诉记者,炒槐米成功是由于当农民的时候就炒过,今年又是跟着懂行的亲戚一起干。接下来他还准备炒薄荷油,而目前薄荷油已达180元/公斤。“这个风险很大,要不是以前也做过一些,现在可不敢做。” 但在亳州市场上,金银花却门庭冷落。去年甲流时期,金银花价格曾高涨一时,甚至达到过400元/公斤的天价。而如今,河北货仅140元/公斤,山东货180元/公斤,较好的河南货也已回落到265元/公斤。 当记者在一个摊位表示要买入金银花作囤货时,看摊的老大爷显得很惊讶,“很少有人囤啦,明年会不会涨,这个谁也说不好。” 明年行情难判断 在药通网的历年数据记录上,记者看到,亳州的四大药之一白芍,往年一直维持3元/公斤的价格。药通网主编郑智文回忆,2003年时,每亩白芍种植成本为800元~1000元,而按照1亩收成800公斤来算,每亩收入为1500元,加上中药材种植对人力要求很高,多年来利润都很低,农民没有种植积极性,五年一收的生长周期,更使得其存量逐年下降。终于在今年供求关系骤紧,使得白芍一跃达到19元/公斤的价格。 而亳州一家大型中药企业的一位采购员则认为,产地气候原因是推高价格的直接因素。如今年甘肃大旱,党参价格从20元/公斤上涨50%;云南先旱后涝,三七价格疯涨至400元/公斤;山西也是先旱后涝,使得山萸肉价格从30元/公斤升到80~90元。不过,“价再高也得采购,每年的库存量是固定的,一定要达到。”据上述企业一名采购经理介绍,公司正在兴建更大的仓库以应对突如其来的药材短缺。 与此同时,近年来中药材的“利好”消息也不断。亳州市地产中药材栽培技术研究所总经理邢振杰就观察到,自今年8月9日“超级细菌”传闻一起,药材价格“一夜之间就涨了”。比如太子参从20元/公斤涨至70元/公斤并一路走高至300元/公斤,而用于抗病毒的桔梗也从60元/公斤涨到90元/公斤。 在这种形势下,一些大企业近年来逐步囤积中药材原料,进一步促成供求关系紧张。今年业内大户康美药业就受益于此轮三七价格高涨,交出漂亮的财报,三季报显示净利润增加60.79%。邢振杰认为,大企业的囤货也对价格上涨起到推波助澜的作用。 游资炒作则被认为是起到了推波助澜的作用。有业内人士表示,中草药产地单一、收获季节集中,产量不大,炒家不用耗费太多的资金就能控制单一药材的价格,容易形成炒作。 但药材价格上涨过快也给不少药厂的生产带来困扰。据东广新闻台报道,上海雷允上药业公司相关人士承认,因中药原料价格上升成本倒挂,已减产板蓝根颗粒和感冒退烧颗粒两款中药。上海中药行业协会相关负责人坦言,很多上海药厂已停产部分中药饮片。 暴涨的价格激发更多的人想分一杯羹。最近一段时间,来邢振杰所在单位拜访的人络绎不绝,多数是来讨教该种何种中药材。一名三十来岁的山西妇女找上门,称自己有近千亩地准备种中药材。另一位山东男子也找到邢振杰,表示将用200亩地种植中药材。“一看有利可图,什么人都来了。”邢振杰笑着说道。 像邢振杰、郑智文这样的内行,身份就如同证券界的分析师,但对于中药材的高风险,仍显得十分谨慎。“预测可真说不好!”郑智文表示,尽管今天高价吸引种植商进入,但并不表明药材价格会迅速回调,因为不少药材是多年生植物,增产效应在短时期内没法显现。 邢振杰则认为,随着流行性疾病的升温,需要密切注意各种消息,可要他说说明年价格还会不会走高,他摇摇头说道:“太复杂了,谁知道呢。”







 我要推广仪器
我要推广仪器
 下载APP
下载APP