2010年版药典氟哌啶醇片红外鉴别方法是:取本品粉末适量(相当氟哌啶醇约50mg),用20ml三氯甲烷分次研磨使溶解,过滤,取滤液水浴蒸干,残渣减压干燥后,压片,与标准图谱比较. 因为氟哌啶醇易溶于三氯甲烷,我在做的过程中就没有剥片;另氟哌啶醇对光 热具不稳定性,滤液采用60度水浴蒸至近干,采用药典原料项下的方法干燥:即60度减压干燥,约5小时,但取出压片时发现它的性状为半固体,压出的片透明度不好,还粘模具,没法做! 会不会是辅料有影响呢?想请教一下,如何改进我的试验,压制一个好的片子?
哌啶和乙醇
现在我们公司增加辅料乙醇和醋酸钠的红外检测,需要购买红外乙醇对照品和醋酸钠对照品。请各位大侠提供除了中检所外,能够在一个星期内买到的厂家。谢谢大家了!!!!
春节好 求助氮氧自由基哌啶醇含量化验方法 谢谢!
请问老师配苍术素对照品时,用甲醇为什么溶解不了,一直有悬浮的颗粒
请问配苍术素对照品时,用甲醇为什么溶解不了,一直有悬浮的颗粒
本人要测定一样品中的哌啶含量,以哌啶纯品进样时出峰,以DMF做纯品的溶剂配成1mg/ml溶液时,哌啶不出峰,DMSO作溶剂时情况一样,但在大约1ml的DMF中加入两三滴纯哌啶时,哌啶出峰,而且峰面积不少,大概换算到1mg/ml的浓度也应该可以检出(1滴大概4~5mg),请问为什么会这样呢?大家有检过哌啶吗,用的是什么溶剂呢?我的检测条件是DB-624,进样口200度,分流比10:1,恒流2ml/min,程序升温40度(5min),15°C/min,220(5min),检测器FID250度,载气是氮气,请大家指教下!
中检所提供的乙醇对照品乙醇浓度是多少?
问下:吸取的对照品无水甲醇、乙醛、乙缩醛是专门的对照品吗?还有用到的50μl、100μl、150μl取样管是微量进样器吗?
安谱提供氯氟醚菊酯对照品: CDAA-CRM352271-250mg 氯氟醚菊酯,99% CAS:352271-52-4主要用于蚊香中氯氟醚菊酯含量的检测
我不小心那错了试剂,对照品用甲醇溶解,我拿了分析纯溶解,是否会影响结果?
本人在岛津GC-2014上做样,FID检测器, 进样口:200,检测器:250,此样品以乙醇为溶剂,溶解以样品限度为浓度的吡啶0.004mg/ml 、哌啶0.004mg/ml、,在DB-624上做过,柱长:30m*0.32mm*1.80um, 进样:1.0ul 以40°C,8min,以20°每分钟升到200°C,保持3分钟,分流20:1、 10:1、5:1都测试过了,可哌啶就是不出峰,从浓溶液定位的图谱来看,哌啶和吡啶出峰时间相差0.1min,其实也就是难以分离,为此换hp-5柱子定位,可是哌啶和吡啶的峰总是拖尾,出峰时间虽然相差0.4min,但是实际按照以上的分流条件做样时候,哌啶不出峰,若是再修改分流比恐怕乙醇过载,杂质变大,更不好判断,于是又换了DM-1色谱柱,同样条件,浓溶液定位,这次哌啶在这柱子上根本不出峰,大浓度也不出峰,无奈又换了DM-WAX柱子做样,同样条件,同样定位,吡啶倒是峰形很好,可哌啶,居然引得基线上升,然后又持平,像是斜坡的样子,但是根本不积分!所以,本人想请问,各位兄台,各位高手,有没有一根柱子是对哌啶响应值高,且完美的分离吡啶和哌啶的,请指教!
请教各位:在乙醇的气相色谱检验中,对照品溶液制备用到的试剂:甲醇、乙醛、乙缩醛、苯、4-甲基-2-戊醇。这些试剂必须用国家标准品吗?还是能用色谱纯的试剂代替,如果能代替使用,需不需要做相关对比试验,又该怎么进行这些对比试验?谢谢各位大神解答。。。。
求金刚烷胺对照品 批号、纯度、厂商
其实我想这么做的原因就是想节约点成本,因为做西青果药材的,对照品没食子酸用50%甲醇溶解,样品也是用50%甲醇溶解,地榆这个药材也是没食子酸,但是浓度稍微低点,我就想用做西青果的对照稀释一下就好,免得再次称对照,地榆药材是用水处理的,药典上写着是称没食子酸适量,用水溶解,现在里面有甲醇,会影响准确性吗?
[size=4]1.所用对照品(标准品)中检所已经发放提供(可参阅中国药典2005年版二部附录ⅩⅤG),且使用方法相同时,应使用中检所提供的现行批号对照品(标准品),并提供其标签和使用说明书,说明其批号,不应使用其他来源者;如使用方法与说明书使用方法不同(如定性对照品用作定量用、效价测定用标准品用作理化测定法定量、UV法或容量法对照品用作色谱法定量等),应采用适当方法重新标定,并提供标定方法和数据;若色谱法含量测定用对照品用作UV法或容量法,定量用对照品用作定性等,则可直接应用,不必重新标定。 2.申报临床研究时,如中检所尚无供应,为不影响注册进度,可先期与中检所接洽制备和标定,申报时提供标定报告、标签(应标明效价或含量、批号、使用效期)和使用说明书;也可与省所合作标定,申报时提供标准品或对照品研究资料,“说明其来源、理化常数、纯度、含量及其测定方法和数据”;标定有困难时,可使用国外药品管理当局或药典委员会发放的对照品(标准品)或国外制药企业的工作对照品(标准品),进行标准制订和其他基础性研究,但应提供其标签(应标明其含量)和使用说明书,能保证其量值溯源性;也可使用国外试剂公司(如sigma公司等)提供的对照品(标准品),但应提供试剂公司该批对照品(标准品)的检测报告(用作含量测定时,应有确定的含量数据),如为高纯度试剂,提供了国外试剂公司检测报告(用作含量测定时,应有确定的含量数据)时,也可使用,并应能保证其量值溯源性,但申请人应及时与中检所接洽对照品(标准品)的标定事宜,临床研究期间完成此工作。 3.直接申报生产品种,如中检所尚无供应,可参照2中要求进行,并提供相应研究资料,但申请人在标准试行期间应与中检所接洽并完成的标定事宜。 4.对照品(标准品)标定的技术要求: 4.1.创新药物 应说明对照品(标准品)原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱),提供标定方法的研究和验证资料(如与原料药质量研究项下相同,可不再提供)、含量测定数据及经统计分析得到的对照品(标准品)含量结果,并说明进行临床前药学研究、药理毒理学研究所用样品的含量是否用该批对照品(标准品)确定或可用该批对照品(标准品)进行量值溯源。 ●纯度测定方法应选用色谱法,并采用两种以上不同分离机理或不同色谱条件并经验证的色谱方法相互验证比较,同时采用二极管阵列检测器或其它适宜方法检测HPLC法的色谱峰纯度,而后根据测定结果经统计分析确定对照品(标准品)原料的纯度。 ●对于组份单一、纯度较高的药物,对照品(标准品)标定方法宜首选可进行等当量换算、精密度高、操作简便快速的容量法。可根据药物分子中所具有的官能团及其化学性质,选用不同的容量分析方法,但应符合如下条件:(1)反应按一个方向进行完全;(2)反应迅速,必要时可通过加热或加入催化剂等方法提高反应速度;(3)共存物不得干扰主药反应,或能用适当方法消除;(4)确定等当点的方法要简单、灵敏;(5)标化滴定液所用基准物质易得,并符合纯度高、组成恒定且与化学式符合、性质稳定(标定时不发生副反应)等要求。 标定方法的选择要关注如下事项:(1)供试品的取用量应满足滴定精度的要求(消耗滴定液约20ml);(2)滴定终点的判断要明确,提供滴定曲线。如选用指示剂法,应考虑其变色敏锐,并用电位法校准其终点颜色。(3)为排除因加入其它试剂而混入杂质对测定结果的影响,或便于剩余滴定法的计算,可采用“将滴定的结果用空白试验校正”的办法;(4)要给出滴定度(采用四位有效数字)的推导过程。 标定结果要根据3个以上实验室各不少于15组测定结果经统计分析,去除离群值和可疑值后的结果,并报告可信限。 ●如该药物没有可进行等当量换算并符合要求的容量法时,可采用反复纯化的原料,色谱法确定纯度后扣除有关物质、炽灼残渣、水分和挥发溶剂等后的理论含量确定为标准品含量,以此为基准进行对照品(标准品)的换代和量值传递。 ●用于抗生素微生物检定法的第一代基准标准品可参照上述方法标定,如为多组份抗生素,其组份比例应与拟上市产品组份比例一致或接近,或以其中某一组份纯品为基准标准品,但要注意标准品换代时量值传递的恒定。 ●仅用于鉴别定性的化学对照品,注重其结构确证的研究资料,纯度和含量的要求一般可适当降低。 ●杂质对照品,用作限度要求时,应提供其来源(合成路线)、结构确证的研究资料,应具备较高的纯度和含量,并提供纯度和含量的的测定结果,提供质量控制标准。 4.2其他类别药物,可参照4.1要求进行。 ●用于抗生素微生物检定法的标准品须用上市国的国家标准品或原发厂的工作标准品为基准标准品进行标定。标定时采用的原料药应符合相应要求,并提供原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱)。标定须用现行版中国药典附录收载的“抗生素微生物检定法”-三剂量法,并提供详细的方法学研究,包括检定菌和培养基的选择、剂量和剂距选择、缓冲液选择(如与质量研究项下相同,可不再提供)。每次标定结果均应照“生物检定统计法-量反应平行线测定法(3.3)”法进行可靠性测验及效价计算。按照《药品注册管理办法》,上市药品质量标准所用标准物质均须由中检所负责标定和管理,药品研发过程中,研制单位应注意及时与中检所联系标定事宜,以保证研发工作的连续性。[/size]
4-羟基哌啶用Aglient1260检测,流动相乙腈,甲醇,各种比例都试过,C18的长柱,短柱都用过,怎么都是不出主峰,求助给些线索!
我现在在合成双哌啶醇酯,反应之后无法分离形成的单酯和双酯,想通过滴定来判断单双酯的含量,请问各位大侠有没有什么好的方法!即测定六元环上的仲氨.
4-羟基哌啶溶于甲醇后,只有甲醇峰,样品再浓都没有?跟柱子有关系吗?
有哪位朋友做过哌啶基哌啶的分析,能否跟我联系下,有一些问题想请教谢谢
问题: 各位老师,有人做过北豆根的含量测定吗?对照品蝙蝠葛苏林碱用甲醇很难溶,试了用流动相也几乎不溶,请问有什么好建议吗
各位老师:乙醇用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测挥发性物质时,进对照品a出来的峰拖尾还分不开是什么原因呢[img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012387514_2300_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012529407_1811_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012539217_9202_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012593807_5993_3926493_3.jpeg[/img]
1-BOC-4-哌啶甲酸检测其纯度时DB-624和HP-5严重拖尾,在DB-WAX中无法出峰,请问选择什么色谱柱合适?或有其它的检测方法
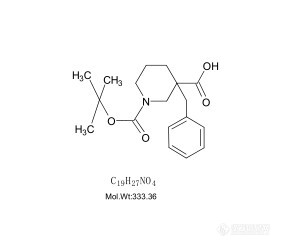
色谱世界的各位大侠们,1-boc-3-苄基哌啶甲酸文献报道说用4.6mm*150mm C18的二氧化硅柱子,用乙腈、水、三氟乙酸作为流动相,在214和254处有吸收峰。我们用紫外全波长扫描后,发现只有在204处有紫外吸收峰,可是我们用乙腈和水作流动相,这个东西在214处不出峰。这个东西该怎么检测纯度呢。这个化合物是通过1-boc-3-哌啶甲酸 和溴苄合成的, 还有1-boc-3-哌啶甲酸 的熔点是159-162℃。 我们这个东西的熔点是109-116℃。 所以这两个东西很定有是不一样的。 实在不行只有打核磁了。我们这个原料的紫外吸收也在204这个位置。但我们用254的紫外薄层检测,发现原料不显色,1-boc-3-苄基哌啶甲酸 轻微显色。[img=,281,247]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231115067664_5937_1815404_3.jpg!w281x247.jpg[/img]
高效液相色谱仪对照品的纯度要求是多少?
【求助】中检所胆固醇对照品要检测胆固醇的含量,但是中检所的胆固醇对照品是用作鉴别的,应该怎么解决这个问题?这样的对照品还能用于含量测定吗?请高手指点,谢谢!
请问各位老师做异丙醇和蔗糖红外鉴别用的什么对照品,中检院的对照品没说可以用于红外鉴别能用吗
请问做HPLC 可以用USP 纯度为91.7%的对照品么?谢谢
各位大神,请教一个小白的问题,关于HPLC中对照品的配制。外标法测定麦芽糖浓度。麦芽糖对照品是99%纯度的一水合物,请教问题是:真空干燥24h后,对照品中的一水合物的水是否会被去除掉,称量的时候是按一水合物的分子量计算来称量,还是按没有水的纯麦芽糖分子量来称量比如要求称1g就在天平称1g?
本人现在用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测硬脂酸镁,为什么对照品溶液中有很多杂峰,我用的正庚烷是色谱纯级别的,还有到最后计算是用不到对照品的浓度,那为什么还要进对照品?请各位老师指点







 我要推广仪器
我要推广仪器
 下载APP
下载APP