化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法1 适用范围 本方法规定了用高效液相色谱法定性检测化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的方法。 本方法适用于化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的定性测定。2 方法提要 样品在经过提取后,经高效液相色谱仪分离,二极管阵列检测器检测,经与平行操作的补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮对照品及补骨脂对照药材比较,以保留时间和紫外光谱图定性,鉴别补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的存在。本方法对补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检出限和取样品0.5 g时的检出浓度见表1。 表1 4种补骨脂特征成分的检出限和检出浓度化合物检出限(ng)检出浓度(μg/g)补骨脂素0.30.6异补骨脂素0.30.6新补骨脂异黄酮0.30.6补骨脂二氢黄酮0.30.63 试剂和材料 除另有规定外,所用试剂均为分析纯,水为实验室用一级水。3.1 乙腈,色谱纯。3.2 补骨脂素,纯度≥99%。3.3 异补骨脂素,纯度≥99%。3.4 新补骨脂异黄酮,纯度≥98%。3.5 补骨脂二氢黄酮,纯度≥99%。3.6 补骨脂,中国食品药品检定研究院,供鉴别用。3.7 补骨脂特征性成分混合标准溶液(=0.1 μg/mL):分别称取补骨脂素(3.2)、异补骨脂素(3.3)、新补骨脂异黄酮(3.4)、补骨脂二氢黄酮(3.5)对照品各5 mg(精确到0.1 mg),置500 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,配制成质量浓度各为10 μg/mL的标准溶液。精密量取各标准溶液0.1 mL置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得0.1 μg/mL的混合标准溶液。3.8 补骨脂标准储备溶液:取补骨脂对照药材0.2 g,置50 mL三角瓶中,加30 mL 70%乙醇回流提取1h,滤过,滤液置100 mL量瓶中,加70%乙醇稀释至刻度,摇匀,即得。4 仪器4.1 高效液相色谱仪:具二极管阵列检测器。4.2 分析天平:感量为0.1 mg。4.3 移液器。4.4 涡旋振荡器。4.5 超声波清洗仪(功率不低于200W)。4.6 高速离心机:转速不小于10000 r/min。
[size=5]超临界流体色谱法测定补骨脂中补骨脂素和异补骨脂素含量[/size] 来源: 作者:陆峰,刘荔荔,李玲,吴玉田摘要目的:建立超临界流体色谱法用于测定补骨脂中补骨脂素和异补骨脂素的含量,并研究其影响因素。方法:用改性的超临界C02萃取中药补骨脂,超临界流体色谱法测定其中的香豆素成分(补骨脂素和异补骨脂素)含量。色谱条件:15 cm×1mm×3μm氨基柱,流动相为含5%甲醇的CO2,柱温40℃,柱头压27.6MPa,UV 247 nm检测。结果:在选定固定相条件下.流动相对组分的洗脱和选择性影响最大,柱温、柱压次之。补骨脂素的回收率为96.9%(RSD=1.8%),异补骨脂素的回收率为95.I%(RSD=I.6%)。培论:与超临界流体萃取法联用,本法可用于香豆素类化合物的分离分析。关键词 超临界流体色谱法;补骨脂素;异补骨脂素;超临界流体萃取法材料和方法 药品和试剂:补骨脂药材由上海长海医院提供,并由本院生药教研室李彬鉴定;补骨脂素(psoralen)、异补骨脂索(isopsoralen)对照品购自中国药品生物制品检定所;SFE和SFC所用CO2购自上海BOC气体公司;蒽(内标物)和氯仿为分析纯;甲醇为HPLC级。 仪器:SFX 2-10萃取器,model 100DX/DM 注射泵,泵控制器. MWD检测器(ISCO.USA),eppendof CH.30色谱柱加热器。 SFE条件:萃取溶剂为CO2,加入氯仿0.06 ml作改性剂,压力为38.5 MPa,温度为70℃:,静态萃取1 min,动态萃取7ml,限流管温度80℃,甲醇作吸收溶剂。 SFC条件:用SpheriorbNH2 柱(150mm×lmm×3μm,A1ltech,USA),流动相为含5%甲醇的CO2.柱头压27.6 MPa,柱温4O℃ ,进样量为0.5, 流速为0.10ml/min,采用50cm×15μm石英毛细管作限流管,检测器AUFS 0.05,247 nm检测。结果与讨论1 固定相/流动相对保留的影响 以超临界C02(含少量改性剂)为基本流动相的SFC是正相色谱,一些极性较弱的化合物在C18柱上几乎不保留 本实验中补骨脂素和异补骨脂素很快从Cl8柱上洗脱,几乎没有分离效果,所以未用C18柱而改用正相NH2柱。 改性剂浓度对组分的保留影响很大。在NH2柱上.用纯C02作流动相时,补骨脂素和异补骨脂素在60min内未洗脱;而在高浓度改性剂时,因两者结构较相似,几乎同时洗脱。本实验用5% 甲醇时洗脱效果良好.2 温度、压力对SFC的影响 温度和压力对保留也有影响。本实验在20.7~ 34.5 MPa压力范围和3O~6O℃ 温度范围内考察了这两个因素的影响。保留时问随压力增大而缩短;随温度升高而延长.3 标准曲线 在上述最佳色谱条件下.补骨脂素、异补骨脂素、内标蒽保留时间分别为6.37,5.00和4.00 min。 用甲醇分别配制对照品溶液和内标溶液:补骨脂素0.423mg/ml,异补骨脂素0.17mg/ml,蒽1.343 mg/ml。分别精密吸取对照品溶液25,50.100.200,400和600μl于10m1量瓶中,加入内标溶液5Oμl,定容,进行SFC分析。以对照品峰与内标峰面积之比(Y)对各自浓度(x,μ/m1)回归.得回归方程。 补骨脂素:Y=23.49X+1.37×10-4,r =0.999; 异补骨脂素:Y:20.04X+0.016.r =0.999。4 方法回收率 精密量取补骨脂素、异补骨脂素溶液各150.250和350μl,加入酸洗硅藻土50mg,挥干溶剂.按SFE条件萃取;甲醇吸收液再加入内标溶液50μl,定容、稀释,各进祥3次,按SFC步骤渊定,计算上述3种浓度的回收率,补骨脂素的回收率为96.9%( r=9.RSD=1.8%);异补骨脂素的回收率为95.1% ( r=9,RSD:1.6% )。5 样品测定 分别取3批干燥药材粉碎.过1OO目筛.精密称取粉末50mg按上述条件萃取;在甲醇吸收液中加入适量内标液,稀释后每份溶液进样3次.按上述色谱条件测定,代入标准曲线,得3批药材中的补骨脂素、异补骨脂索含量分别为0.77%.0.70% ;0.86% .0.82% :0.65% ,0.53% 。 以上结果表明:流动相组成是影响分离的最重要因素,加入改性剂可大大改善中等极性化合物的洗脱分离。系统的温度和压力对分离也有影响。与SFE联用,SFC可以较方便地用于某些香豆素类化合物的分离分析。
化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法

作者:程龙琼, 周晓英(成都市食品药品监督检测中心,四川 成都 610045)摘要:目的采用HPLC法测定青娥丸中补骨脂的含量。方法色谱柱为钻石C18柱(200 mm×4.6 mm,5μm)。流动相为甲醇-水(55:45);流速1.0 ml.min-1;检测波长246 nm;柱温为室温。结果补骨脂素的线性范围是0.1004~1.5060μg(r=0.9998)。平均加样回收率为97.8%,RSD=1.92%(n=6)。异补骨脂素的线性范围是0.0830~1.2450μg(r=0.9993)。平均加样回收率为98.5%,RSD=1.60%(n=6)。结论所建方法可用于测定青娥丸中的补骨脂。谱图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207161015_377764_1606903_3.jpg
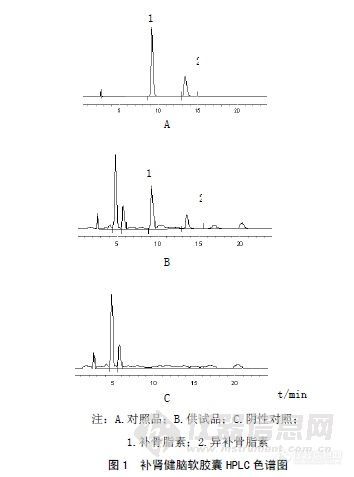
作者:徐晓明;徐大勇;邢俊波;(梅河口市卫生职工中等专业学校;梅河口市医院爱民医院药剂科;中国人民解放军总后卫生部药品仪器检验所;)摘要:目的建立补肾健脑软胶囊中补骨脂素和异补骨脂素的含量测定方法。方法采用HPLC法,色谱柱为Diamonsil-C18(250mm×4.6mm,5μm),流动相为甲醇-水(48∶52),流速为1.0mL/min,检测波长为246nm,柱温40℃。结果补骨脂素和异补骨脂素线性范围分别为0.01008~0.252μg(r=0.9998)、0.00914~0.2285μg(r=0.9999)。平均回收率分别为98.91%(RSD=1.74%)、99.93%(RSD=1.27%)。结论本法分离度好,快速、简便,可作为该产品的质量控制方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208131425_383490_1606903_3.jpg
[b][size=16px]药用补骨脂[/size][/b][size=16px][/size][b][size=16px]《药性论》:“主男子腰疼,膝冷囊湿,逐诸冷痹顽,止小便利,腹中冷。”《本草纲目》:“治肾泄,通命门,暖丹田,敛精神。”《本草求真》:“能敛神明,使心包之火与命门之火相通,因而元阳坚固,骨髓充实。”《本草经疏》:“能暖水脏,阴中生阳,壮火益土之要药也。”[/size][/b][size=16px][/size][size=16px]补骨脂单用有效,也常与杜仲、核桃仁、熟地黄、五味子、菟丝子等药材同用,以增强补肾壮阳、固精缩尿的效果。[/size][size=16px][/size][size=16px]如用于治疗由肾阳不足引起的阳痿遗泄、尿频、遗尿等症,常配伍仙灵脾、菟丝子等同用。治虚冷泄泻,常与肉豆蔻等同用。肾气不足,摄纳无权,引起喘促,补骨脂温肾而纳气平喘,常与胡桃肉配伍以治虚寒气喘。[/size]
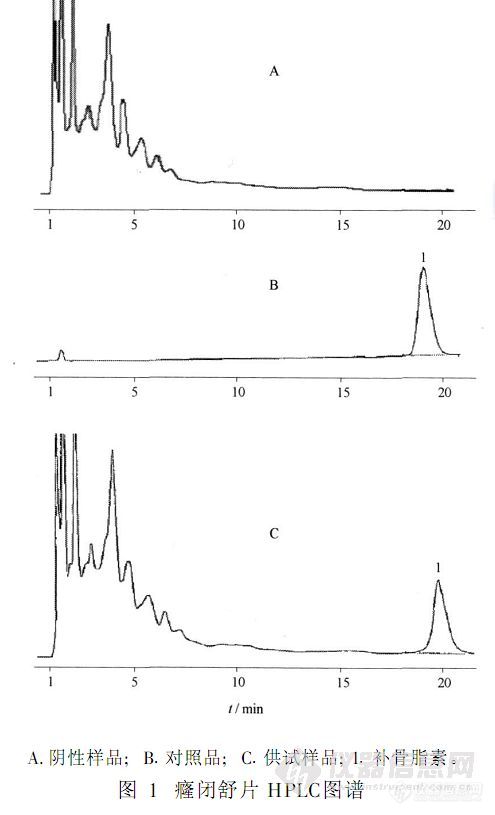
【作者】 叶英响;【机构】 浙江省义乌市中心医院; 温州医学院附属义乌医院中药科;【摘要】 目的:建立癃闭舒片中补骨脂素的含量测定方法;方法:采用高效液相色谱法,色谱柱Diamonsil C18(4.6 mm×250 mm,5μm),流动相甲醇-1%冰醋酸水溶液(45∶55),检测波长297 nm;结果:补骨脂素回归方程为Y=41 651X+9 228,r=0.999 4,线性范围为3.52~17.6μg.mL-1,平均回收率101.1%,RSD 2.1%。结论:本方法简单、稳定,测定准确。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208131339_383473_2379123_3.jpg
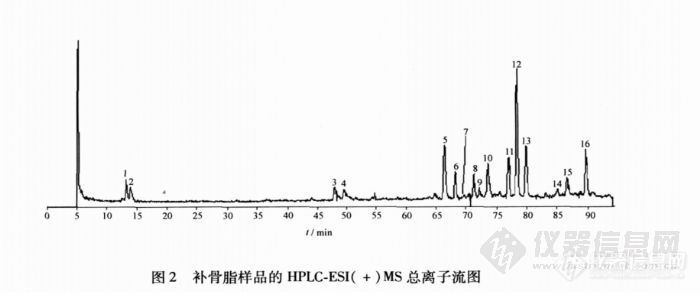
【作者】 刘亚男; 王跃飞; 韩立峰; 潘桂湘; 王虹;【Author】 LIU Ya′nan,WANG Yuefei,HAN Lifeng,PAN Guixiang,WANG Hong(Engineering Research Center of Modern Chinese Medicine Discovery and Preparation Technique Traditional Chinese MedicineResearch Centre of Tianjin University of Traditional Chinese Medicine,Tianjin 300193,China)【机构】 天津中医药大学中医药研究院现代中药发现与制剂技术教育部工程研究中心;【摘要】 目的:建立补骨脂药材中化学成分的高效液相色谱-电喷雾质谱分析方法。方法:采用DiamonsilTMC18(4.6 mm×250 mm,5μm)色谱柱;流动相0.05%甲酸水溶液-乙腈,梯度洗脱;流速1 mL.min-1;柱温30℃;DAD扫描范围190~400nm;检测波长246 nm。Finnigan电喷雾离子阱多级质谱仪;正离子检测模式;ESI喷雾电压4 500 V;鞘气(N2)流速60个单位;辅助气(N2)流速20个单位;毛细管温度350℃;毛细管电压19 V,扫描范围m/z90~800。结果:补骨脂中化学成分获得了较好的分离和检测,共鉴定出2个香豆素苷,3个香豆素,8个黄酮和1个单萜酚类成分。结论:该方法灵敏度高、分离度好,适用于补骨脂药材中化学成分的快速定性鉴定。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301609_380605_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207301609_380606_2379123_3.jpg
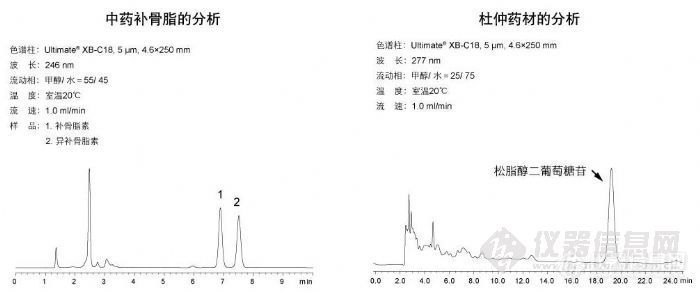
中药 中药补骨脂的分析和杜仲中松脂醇二葡萄糖苷的分析http://ng1.17img.cn/bbsfiles/images/2009/10/200910311054_179312_1896702_3.jpg
这个问题可能有些奇怪先说说概况吧:之前我们这边的台帐都是采用活页装订的,就是缺少了可以直接拆了往里面加页。上次检查被专家提出这样有造假嫌疑,建议我们采用固定页数受控发放的台帐。http://simg.instrument.com.cn/bbs/images/brow/em09501.gif于是我们就准备这么开始做了,但做到对照品领用这一块的时候就发现问题了。对照品的帐都是一页写一个对照品,而很多对照品领用数量不固定,可能用到1页、2页,甚至可能3页或更多。这样的话,页数就无法固定了。想过一个对照品用一本帐,但我们这边对照品实在是太多了,一个一本的话得一大堆帐http://simg.instrument.com.cn/bbs/images/brow/em09512.gif。不知道各位都是怎么处理对照品台帐的?
[em06] 如题菲罗门柱250*4.6,1.0ml/min,246nm 其中对照品浓度为:补14.32ug/ml,异补10.064ug/mlyy000021 对照品 柱温35度 进样20ul 两峰分离度 2.41 补塔板数4023 甲醇:水(43:57)yy000022 对照品 35度 进样20ul 两峰分离度 2.37 补塔板数4015 甲醇:1%醋酸(43:57)yy000024 对照品 40度 进样5ul 两峰分离度 3.19 补塔板数8117 甲醇:1%醋酸(43:57)yy000025 中药复方制剂的醇提液 35度 进样10ul 两峰分离度 3.13 补塔板数7415 甲醇:1%醋酸(43:57)

茵栀解毒颗粒 绿原酸对照品色谱图,出峰时间在9到25分钟之间随意变换 , 不固定 ,求解决方案
对照品溶液的制备 取补骨脂素对照品和异补骨脂素对照品适量,精密称定,加甲醇制成每1ml各含2ug的溶液,即得。供试品溶液的制备 取本品水蜜丸,研细,取0.5g,精密称定,置研钵中,加硅藻土2g,研匀,转移至具塞锥形瓶中,精密加入甲醇25ml,密塞,称定重量,超声处理(功率250W,频率50kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。问题1、对照品溶液制备的过程中,补骨脂素对照品和异补骨脂素对照品是称好放同一量瓶中溶解稀释更好,还是分别配好呢?2、这个供试品的制备中,精密称定后,“置研钵中,加硅藻土2g,研匀,转移至具塞锥形瓶中”,我觉得这个精密称定后,加硅藻土置研钵中研匀会造成样品的损失吧,你再怎么倒也倒不干净吧。。。。我是直接称好放入锥形瓶中,再加2g硅藻土进去,拿个玻璃棒搅一下,不知道这样可不可以???
对照品溶液与供试品溶液需使用相同的顶空条件,因为样品瓶中有气-液或气-固两相,甚至气-液-固三相共存。顶空气体中各组分的含量既与其本身的挥发性有关,又与样品基质有关。特别是那些在样品基质中溶解度大(分配系数大)的组分,“基质效应”更为明显。这是顶空进样的一大特点,即顶空气体的组成与原样品中的组成不同,这对定量分析的影响尤为严重。因此,对照品不能仅用待测物的标准品配制,还必须有与原样品相同或相似的基质,否则,定量误差将会很大。实际应用中一些消除或减少基质效应的方法:a、利用盐析作用即在水溶液中加入无机盐(如过饱和硫酸钠溶液)来改变挥发性组分的分配系数;b、在有机溶液中加入水相,这可以减小有机物在有机溶剂中的溶解度,增大其在顶空气体中的含量;c、调节溶液的pH 对于碱和酸,通过控制pH可使其解离度改变,或使其中待测物的挥发性变得更大,从而有利于分析;d、降低样品浓度也是减小基质效应的常用方法,但其代价是减低了灵敏度;e、固体样品中挥发物的扩散速度很慢,往往需要很长时间才能达到平衡。尽量采样全溶的样品溶液,有利于缩短平衡时间;f、其他消除基质效应的技术,如全挥发技术等
请教各位大侠:标准品(对照品)怎样定值?具体说来:1. 用HPLC对X物质(自制标准品)进行纯度测定时,测定的是X物质和2,3-二氨基萘(一种衍生试剂)的衍生物质,这种情况下,对X物质(标准品)如何定值2. 衍生试剂对X(标准品)物质有干扰如何排除?(在保证衍生试剂过量的情况下,增加衍生试剂浓度,测定X物质峰面积增大(按理说,X物质和衍生试剂完全反应,这时,测定衍生后的X物质峰面积应该不会变化才对啊)3. 常用什么方法(除归一法外)?单位目前正在开展X标准品研制计划工作,请问各位专家提供宝贵建议,欢迎指导。非常感谢。
有一个自制的多肽类工作对照品(1类新药),目前是用定氮法来测去含量,然后再用于测样品含量。该对照品纯度只有95%左右,也就是说还有5%的未知结构的短肽或缺失肽存在。而定氮法测的总氮,这样算出来的对照品含量应该是高于它的实际含量的,用它测的样品含量也应该是虚高了的。所以我认为用定氮法来测定多肽类的对照品含量不是很准确。请问:我这种理解对吗?如果对,有什么更准确的方法检测多肽产品的含量呢?谢谢[em0801]
如题请问用芦丁对照品绘制标准曲线是否能用槲皮素或其他的黄酮对照品替代?由于实验室的芦丁标准品不见了,老师建议先用槲皮素替代,请问是否可行?
[font=宋体][color=black][back=white]对照品溶液与供试品溶液需使用相同的顶空条件,因为样品瓶中有气[/back][/color][/font][font=宋体][color=black][back=white]-液或气-固两相,甚至气-液-固三相共存。顶空气体中各组分的含量既与其本身的挥发性有关,又与样品基质有关。特别是那些在样品基质中溶解度大(分配系数大)的组分,“基质效应”更为明显。这是顶空进样的一大特点,即顶空气体的组成与原样品中的组成不同,这对定量分析的影响尤为严重。因此,对照品不能仅用待测物的标准品配制,还必须有与原样品相同或相似的基质,否则,定量误差将会很大。[/back][/color][/font]
大家判定样品和对照品的红外光谱图一致是怎么描述的?
大家经常做气相需要测对照品溶液,有时对照品溶液的出峰面积在不同时期可能差异较大,大家遇到过这样的问题吗?问题:取甲醇、乙酸乙酯、甲苯适量,精密称定,用DMF溶解稀释定容制成每1ml中约含甲醇60ug、乙酸乙酯100ug、甲苯20ug的混合溶液。那么我们实际称取的对照质量应该在什么范围内是可以接受的?因为称量的差异会导致对照品出峰面积的差异。问题:如何能用天平称准对照试剂的量?(有机溶剂甲醇、乙腈、二氯甲烷等)换算成体积量取还是直接称取?
请问哪里有农残对照品订购,
现在有标准品为进口丁基橡胶作为测定门尼仪测定校正样品,由于进口标准品价格高,现在用国产丁基橡胶与标准丁基橡胶在相同条件下同一台仪器测定,多少次对比数据,如何把测定值和标准品的标准值转换成对照品的标准值.
对照品:用于鉴别、检查、含量测定和校正检定仪器性能的标准物质;对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。 标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。 对照品与标准品概念不清?对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已,造成错误的原因,可能是有的药品既有对照品,又有标准品。 例如:当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品。即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的。
[size=3]不知大家注意没有,在2010年版药典中,特别是UV-Vis测含量,在“对照品溶液的制备”中,往往是准确指出精密称取的对照品的量,例如,2010年版药典一部第5页,人工牛黄中胆酸的含量测定项下,胆酸对照品溶液的制备:取胆酸对照品12.5mg,精密称定,置25ml量瓶中,加60%冰醋酸溶液使溶解,并稀释至刻度,摇匀,即得(每1ml中含胆酸0.5mg)。而,HPLC或GC等测含量,大多的表述是,如同是第5页,八角茴香中反式茴香脑的含量测定,对照品溶液的制备:取反式茴香脑对照品适量,精密称定,加乙醇制成每1ml含0.4mg的溶液,即得。这两种表述有何不同?[/size]
请问下大家,对照品配制你们是一次同时配两个浓度,做一下校正因子,还是和以前没用完的校正啊?一般两个对照品的RSD应该控制在多少?
化验室有一台低温柜(-25℃),用来放置药品标准品和对照品,做仪器设备确认需做哪些项目?标准如何定?有检定规程、规范或者国标之类的规定可参考吗?了解的帮忙回答下,谢谢。
[size=3]新的2010年版药典一部中,特别是药材所用的对照品,无论是鉴别,还是含量测定,都多了不少陌生的对照品,名称也很奇怪。这些对照品是怎样制得的?如果是从原植物中分得的,那得用多少的植物?成本得有多高![/size]
请教一下:薄层鉴别用的对照品溶液一般都是说每1ml含1mg的对照品溶液,这个具体怎么配制的一个操作步骤?需要定量定容的吗?谢谢各位老师
[font=&][color=#333333]对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。[/color][/font][font=&][color=#333333]标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单λ(U)表示。[/color][/font][font=&][color=#333333]如果还是感觉不甚明了,是否标准品只用于生物方面?是否化学方面只能称对照品?标准品有什?要求?对照品有什?要求?[/color][/font][font=&][color=#333333]国家药品标准品、对照品系指国家药品标准中用于鉴别、检查、含量测定、杂质和有关物质检查等标准物质,它是国家药品标准不可分割的组成部分。国家药品标准物质是国家药品标准的物质基础,它是测量药品质量的基准;也是做为校正测试仪器与方法的物质标准;在药品检验中,它是确定药品真α优劣的对照,是控制药品质量必不可少的工具。[/color][/font][font=&][color=#333333]目前,中国药品生物制品检定所已能提供各类国家标准物质1242种,其中中药化学对照品288种,对照药材400种,两者占总数的一半以上。[/color][/font][font=&][color=#333333]国家标准品及生物参考品系指用于鉴别、检查含量或效价测定的标准物质,其制备与标定应符合“生物制品国家标准物质制备和标定规程”要求,并由国务院药品监督管理部门指定的机构分发。企业工作标准品或参考品必须经国家标准品或参考品标化后方能使用。[/color][/font][font=&][color=#333333]对照品系指用于生物制品理化等方面测定的特定物质,由生产单λ采用与制品生产工艺相同的方法制备。对照品应尽可能与制品原液配方一致,稳定性较差的,可加不含对测定有干扰物质的适宜的稳定剂。对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。[/color][/font][font=&][color=#333333]标准品、对照品:是指用于鉴别、检查、含量测定的标准物质,均由国务院药品监督管理部门指定的单λ制备、标定和供应。标准品系指用于生物测定、抗生素或生化药品中含量或效价测定的标准物质,一国际标准品进行标定;对照品出另有规定外,按干燥进行计算后使用。[/color][/font][font=&][color=#333333]标准品和对照品均附有使用说明书,质量要求,有效期和装量等。[/color][/font][font=&][color=#333333]生物制品标准物质系指用于生物制品效价、活性或含量测定的或其特性鉴别、检查的生物标准品或生物参考物质。[/color][/font]转自:食品伙伴网
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo







 我要推广仪器
我要推广仪器
 下载APP
下载APP