如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
我自己翻了,希望能有更专业的帮忙翻译,我自己对照学习氧 氟 沙 星 片Yangfushaxing PianOfloxacin Tablets本品含氧氟沙星(C18H20FN3O4)应为标示量的90.0%~110.0%。【性状】 本品为类白色或微黄色片或薄膜衣片,除去包衣后显类白色至微黄色。【鉴别】 (1)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。(2)取本品细粉适量,用0.1mol/L盐酸溶液溶解并稀释制成每1ml中含氧氟沙星6μg的溶液,滤过,滤液照紫外-可见分光光度法(附录Ⅳ A)测定,在294nm的波长处有最大吸收。【检查】 有关物质 取含量测定项下的供试品贮备液作为供试品溶液;精密量取适量,加0.1mol/L盐酸溶液稀释成每1ml中含6μg的溶液,作为对照溶液。照氧氟沙星有关物质项下的方法测定,供试品色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积(0.5%),各杂质峰面积的和(任何小于对照溶液主峰面积的0.05倍的峰可忽略不计)不得大于对照溶液主峰面积的2倍(1.0%)。溶出度 取本品,照溶出度测定法(附录Ⅹ C 第一法),以盐酸溶液(9→1000)900ml为溶出介质,转速为每分钟50转,依法操作,经30分钟时,取溶液适量,滤过,精密量取续滤液2ml,置50ml量瓶中,加溶出介质稀释至刻度,摇匀,照紫外-可见分光光度法(附录Ⅳ A),在294nm的波长处测定吸收度;另取氧氟沙星对照品适量,精密称定,加溶出介质溶解并稀释成每1ml中约含4.5μg的溶液,同法测定,计算每片的溶出量。限度为标示量的80%,应符合规定。其他 应符合片剂项下有关的各项规定(附录Ⅰ A)。【含量测定】照高效液相色谱法(附录Ⅴ D)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300使溶解,用磷酸调节pH值至2.2)-乙腈(85:15)为流动相;检测波长为294nm。取氧氟沙星适量,用0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含1mg的溶液,置紫外光灯(254nm)下照射4小时以上,取10μl注入液相色谱仪,理论板数按氧氟沙星峰计算不低于5000,紧邻氧氟沙星峰前的杂质峰与氧氟沙星峰的分离度应符合要求。测定法 取本品10片,精密称定,研细,精密称取适量(约相当于氧氟沙星0.1g),置100ml量瓶中,加0.1mol/L盐酸溶液溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品贮备液,精密量取5ml,置50ml量瓶中,加0.1mol/L盐酸溶液至刻度,摇匀,精密量取10μl分别注入液相色谱仪,记录色谱图;另取氧氟沙星对照品适量,精密称定,用0.1mol/L盐酸溶液溶解并定量稀释制成每1ml中含氧氟沙星0.12mg的溶 液,同法测定,按外标法以峰面积计算,即得。【类别】 同氧氟沙星。【规格】 0.1g【贮藏】 遮光,密封保存。
高手们,能不能给我指点一下,标准品和对照品有啥区别呢?什么时候用标准品什么时候用对照品好友回复1:标准品现在基本特指生物类的,其他的叫对照品好友回复2:可以有溯源的是标准品,没有溯源的就是对照品大家说说看
氯唑沙宗对照品和对乙酰氨基酚对照品的出峰时间分别是多久?
[b]稀土离子Tb抖能与药物培氟沙星形成1:2络合物,并发出Tb。 的特征荧光,加入表面活性剂十二烷基硫酸钠(sDs)能显著增强该体系的荧光强度,据此建立了一种表面活性剂敏化Tb 荧光探针测定培氟沙星的新方法。在pH 5.8~6.8,Tb。 和SDS浓度分别为5.0×10 toolL 和1.0×10 toolL 的条件下,培氟沙星的浓度与体系的荧光强度呈良好的线性关系,线性范围为3.0×10 ~7.9×10 toolL ;检出限为3.0×10 toolL 。该方法可用于培氟沙星制剂和血清中药物含量的直接测定。[/b]
请问老师配苍术素对照品时,用甲醇为什么溶解不了,一直有悬浮的颗粒
请问配苍术素对照品时,用甲醇为什么溶解不了,一直有悬浮的颗粒
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo
在检测注射用甲磺酸左氧氟沙星含量时:中间体检测时用紫外检测,成品用的是高效液相检测,之前中间体用原料做工作对照,对照品A值一般在所配浓度下一般为0.39左右,F值在1.26左右,成品没什么问题,可是现在用中检所的对照品,中间体测定时对照品在相同浓度下A值才有0.35左右,这样的话F值就接近1.3了,结果含量就会高很多甚至不合格,但是成品上液相时又是合格的,也就是说对照品应该没问题,是不是中检所的对照品不适合用紫外分光光度法来检测呢?请各位帮忙分析分析,谢谢!!!补充:在这之前,该产品中间体检测都是用中检所的对照品上高效液相测定的,为了节省时间改用了紫外分光光度法检测工艺没改,液相测出来接过肯定会更精确些,但是奇怪的是,同样的对照品中间体检测超标,成品检测合格,不过之前用原料做工作对照检中间体时,成品标示量一般在100%以下,用中检所对照品检测中间体,成品标示量一般在105%,规定上限是110%没我是在想我们的中间体检测方法是不是不太合理制药工艺中没有纯化过程的,但是原料工作对照77.54%,中检所对照品97.3%,都是根据所需浓度换算好了再配制检测的,我是想应该里面所含杂质吸收影响就不明显了吧?您觉得呢?
请教各位大侠,在平时的工作中,用于HPLC的对照品(中药)一般多久配一次?用什么指标衡量该重新配对照了?新配的对照又是用什么指标衡量是否可以取代旧对照了?

http://ng1.17img.cn/bbsfiles/images/2017/01/201701191656_648524_2518341_3.png征集化妆品中诺氟沙星的检测方法相关新闻http://www.woyaoce.cn/news/newsdetails.aspx?id=86131
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7681612594 七氟醚杂质C Sevoflurane Related Compound C 对照品/标准品1612572 七氟醚杂质 B Sevoflurane Related Compound B 对照品/标准品1612550 七氟醚杂质 A Sevoflurane Related Compound A 对照品/标准品1612540 七氟醚 Sevoflurane 对照品/标准品1612539 盐酸舍曲林 Sertraline Hydrochloride 对照品/标准品1612528 盐酸舍曲林杂质A Sertraline Hydrochloride Related Compound A 对照品/标准品1612517 盐酸舍曲林消旋体混合物 Sertraline Hydrochloride Racemic Mixture 对照品/标准品1612506 L-丝氨酸 L-Serine 对照品/标准品1612426 芝麻油杂质B Sesame Oil Related Compound B 对照品/标准品1612415 芝麻油杂质A Sesame Oil Related Compound A 对照品/标准品1612404 芝麻油 Sesame Oil 对照品/标准品1612029 番泻苷 B Sennoside B 对照品/标准品1612018 番泻苷 A Sennoside A 对照品/标准品1612007 番泻苷 Sennosides 对照品/标准品1611955 硒 蛋氨酸 Selenomethionine 对照品/标准品1611900 盐酸司来吉兰 Selegiline Hydrochloride 对照品/标准品1611004 司可巴比妥 CII Secobarbital CII 对照品/标准品1610090 东莨菪亭 Scopoletin 对照品/标准品1610001 氢溴酸东莨菪碱 Scopolamine Hydrobromide 对照品/标准品1609831 沙奎那韦杂质A Saquinavir Related Compound A 对照品/标准品1609829 甲磺酸沙奎那韦 Saquinavir Mesylate 对照品/标准品1609807 双水杨酯 Salsalate 对照品/标准品1609625 沙美特罗杂质B Salmeterol Related Compound B 对照品/标准品1609614 沙美特罗杂质A Salmeterol Related Compound A 对照品/标准品1609603 昔美酸沙美特罗 Salmeterol Xinafoate 对照品/标准品1609501 水杨酸片 Salicylic Acid Tablets 对照品/标准品1609024 水杨酸杂质B Salicylic Acid Related Compound B 对照品/标准品1609013 水杨酸杂质A Salicylic Acid Related Compound A 对照品/标准品1609002 水杨酸 Salicylic Acid 对照品/标准品1608000 水杨酰胺 Salicylamide 对照品/标准品1607506 连翘粉状贯叶提取物 Powdered St. John's Wort Extract 对照品/标准品1607040 糖精钠 Saccharin Sodium 对照品/标准品1607029 糖精钙 Saccharin Calcium 对照品/标准品1607007 糖精 Saccharin 对照品/标准品1606503 芦丁 Rutin 对照品/标准品1606208 硝酚胂酸 Roxarsone 对照品/标准品1605523 罗哌卡因杂质B Ropivacaine Related Compound B 对照品/标准品1605512 罗哌卡因杂质A Ropivacaine Related Compound A 对照品/标准品1605500 盐酸罗哌卡因 Ropivacaine Hydrochloride 对照品/标准品1604916 罗库溴铵合剂峰的识别 Rocuronium Peak Identification Mixture 对照品/标准品1604905 罗库溴铵 Rocuronium Bromide 对照品/标准品1604870 利凡斯的明杂质B Rivastigmine Related Compound B 对照品/标准品1604869 利凡斯的明杂质A Rivastigmine Related Compound A 对照品/标准品1604814 利托那韦杂质混合物 Ritonavir Related Compounds Mixture 对照品/标准品1604803 利托那韦 Ritonavir 对照品/标准品1604701 盐酸利托君 Ritodrine Hydrochloride 对照品/标准品1604665 利培酮系统适用性试验用混合物 Risperidone System Suitability Mixture 对照品/标准品1604654 利培酮 Risperidone 对照品/标准品1604643 利塞膦酸杂质C Risedronate Related Compound C 对照品/标准品1604632 利塞膦酸杂质B Risedronate Related Compound B 对照品/标准品1604621 利塞膦酸杂质A Risedronate Related Compound A 对照品/标准品1604610 利塞膦酸钠 Risedronate Sodium 对照品/标准品1604600 利美索龙 Rimexolone 对照品/标准品1604508 盐酸金刚乙胺 Rimantadine Hydrochloride 对照品/标准品1604348 利鲁唑杂质A Riluzole Related Compound A 对照品/标准品1604337 利鲁唑 Riluzole 对照品/标准品1604202 醌式利福平 Rifampin Quinone 对照品/标准品1604009 利福平 Rifampin 对照品/标准品1603800 利福布丁 Rifabutin 对照品/标准品1603108 核糖 Ribose 对照品/标准品1603006 维生素B2 Riboflavin (Vitamin B2) 对照品/标准品1602706 利巴韦林 Ribavirin 对照品/标准品1602003 间苯二酚 Resorcinol 对照品/标准品1601849 二类残留溶剂-二甲苯 Residual Solvent Class 2 - Xylenes 对照品/标准品1601827 二类残留溶剂-三氯乙烯 Residual Solvent Class 2 - Trichloroethylene 对照品/标准品1601805 二类残留溶剂-甲苯 Residual Solvent Class 2 - Toluene 对照品/标准品1601780 二类残留溶剂-四氢萘 Residual Solvent Class 2 - Tetralin 对照品/标准品1601770 二类残留溶剂-四氢呋喃 Residual Solvent Class 2 - Tetrahydrofuran 对照品/标准品1601769 二类残留溶剂-二氧噻吩烷 Residual Solvent Class 2 - Sulfolane 对照品/标准品1601747 二类残留溶剂-吡啶 Residual Solvent Class 2 - Pyridine 对照品/标准品1601725 二类残留溶剂-硝基甲烷 Residual Solvent Class 2 - Nitromethane 对照品/标准品1601703 二类残留溶剂-N-甲基吡咯烷酮 Residual Solvent Class 2 - N-Methylpyrrolidone 对照品/标准品1601689 二类残留溶剂-甲基环己烷 Residual Solvent Class 2 - Methylcyclohexane 对照品/标准品1601667 二类残留溶剂-甲基丁基酮 Residual Solvent Class 2 - Methylbutylketone 对照品/标准品1601645 二类残留溶剂- 2-甲氧基乙醇 Residual Solvent Class 2 - 2-Methoxyethanol 对照品/标准品1601623 二类残留溶剂-甲醇 Residual Solvent Class 2 - Methanol 对照品/标准品1601601 二类残留溶剂-己烷 Residual Solvent Class 2 - Hexane 对照品/标准品1601587 二类残留溶剂-甲酰胺 Residual Solvent Class 2 - Formam
昨天用Agilent1200测定盐酸左氧氟沙星胶囊的含量,自动进样,流动相是在线混合。一份对照连续进样5次,保留时间基本一致,但峰面积变化很大,RSD=6.25%,测定结果偏差很大,不能使用。是进样器出了问题,还是流动相混合有问题,或者别的什么原因?
《水和废水监测分析方法》四版中的“水中的细菌学测定”有提到要对培养基进行“阳性和阴性对照培养检查试验”,举个例子(摘自:表 5-2-4): 对照培养细菌类别 阳性 阴性粪大肠菌类 大肠埃希氏菌 产气肠杆菌是不是说要在培养基中分别加入大肠埃希氏菌和产气肠杆菌,看它们是否分别呈现阳性和阴性反应,如果阳性和阴性反应都符合要求的话培养基就合格?大家讨论一下,谢谢!
对照品的峰型出现这种现象是样品过载吗
[font=宋体][font=宋体]检测农产品中的喹诺酮类药物对于第三方检测食品实验室而言是再熟悉不过的项目了,氧氟沙星作为常检项目之一,主要针对水产品、牛羊等畜肉,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]法检测主要依据国家标准[/font][font=Calibri]GB 31658.17-2021[/font][font=宋体]和农业部[/font][font=Calibri]1077[/font][font=宋体]号公告[/font][font=Calibri]-1-2008[/font][font=宋体]。就这么一个常规项目,在前段时间出现诡异的污染现象。[/font][/font][font=宋体]当时在做一批的水产品检测时,发现[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测结果所有的样品中都有氧氟沙星检出,而且结果均超过了最大残留限值。当时首先考虑的是基质干扰,因为氧氟沙星常出现保留时间接近的干扰峰,查看相对离子丰度比发现确实是氧氟沙星。考虑到空白样品中也有氧氟沙星检出,可能是前处理过程中带入的污染。重新配置了内标工作溶液,并将这批样品换人重做后问题并没有得到解决,因此怀疑污染源在仪器端。[/font][font=宋体][font=宋体]我将第一次处理的样品换到另一台[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]上检测,结果也有氧氟沙星检出,检出值小于之前的结果。分析三次的测试结果发现:所有样品、溶剂空白、样品空白都有氧氟沙星检出,保留时间基本一致,同一台仪器的仪器不同样品之间的结果接近,同一样品不同仪器的检测结果不一致。怀疑污染是在进样过程中引入,连同容器一起更换了新的洗针溶液,充分灌注洗针液通道,多次运行清洗进样针、针密封清洗、注射器清洗程序。用之前的溶剂空白,连续进样[/font][font=Calibri]10[/font][font=宋体]次,测试结果显示,溶剂空白中不再有氧氟沙星检出。将之前换下的洗针溶液取样上机测试,发现两台仪器的弱洗溶液中都有氧氟沙星检出。将之前的水产样品上机测试,结果均未检出氧氟沙星。[/font][/font][font=宋体]在后续查找污染原因时发现,同时在配置洗针溶液时未规范佩戴手套与口罩,同时他最近在服用氧氟沙星片剂。当时配置的洗针液直接添加到仪器洗针液的容器中,剩余部分倒入了另一台仪器的洗针液容器中。[/font][font=宋体]这次污染事件提醒了我们,在使用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]这类高灵敏度的仪器时,规范的实验操作的重要性。实验室防护品不仅保护我们的健康,也是实验结果准确的保障。不同仪器间,避免出现试剂混用的现象,这样当出现问题时,我们才能更准确的找到原因。[/font]
安谱提供氯氟醚菊酯对照品: CDAA-CRM352271-250mg 氯氟醚菊酯,99% CAS:352271-52-4主要用于蚊香中氯氟醚菊酯含量的检测
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7651724918 唑吡坦杂质A CIV Zolpidem Related Compound A CIV 对照品/标准品1724907 酒石酸唑吡坦 CIV Zolpidem Tartrate CIV 对照品/标准品1724893 唑吡坦 CIV Zolpidem CIV 对照品/标准品1724805 盐酸唑拉西泮 Zolazepam Hydrochloride 对照品/标准品1724769 硫酸锌 Zinc Sulfate 对照品/标准品1724747 氧化锌 Zinc Oxide 对照品/标准品1724689 齐留通杂质C Zileuton Related Compound C 对照品/标准品1724678 齐留通杂质B Zileuton Related Compound B 对照品/标准品1724667 齐留通杂质 A Zileuton Related Compound A 对照品/标准品1724656 齐留通 Zileuton 对照品/标准品1724532 齐多夫定杂质C(胸腺嘧啶) Zidovudine Related Compound C (thymine) 对照品/标准品1724521 齐多夫定杂质B Zidovudine Related Compound B 对照品/标准品1724500 齐多夫定 Zidovudine 对照品/标准品1724317 扎西他滨杂质A Zalcitabine Related Compound A 对照品/标准品1724306 扎西他滨 Zalcitabine 对照品/标准品1724000 盐酸育亨宾 Yohimbine Hydrochloride 对照品/标准品1722005 木糖 Xylose 对照品/标准品1721002 盐酸赛洛唑啉 Xylometazoline Hydrochloride 对照品/标准品1720600 木糖醇 Xylitol 对照品/标准品1720429 盐酸赛拉嗪 Xylazine Hydrochloride 对照品/标准品1720407 赛拉嗪 Xylazine 对照品/标准品1720203 呫吨酮 Xanthone 对照品/标准品1720000 呫吨酸 Xanthanoic Acid 对照品/标准品1719102 华法林杂质 A Warfarin Related Compound A 对照品/标准品1719000 华法林 Warfarin 对照品/标准品1717708 牡荆素(牡荆甙) Vitexin 对照品/标准品1717504 含量测定系统适用性用维生素D Vitamin D Assay System Suitability 对照品/标准品1716002 维生素A Vitamin A 对照品/标准品1715000 硫酸紫霉素 Viomycin Sulfate 对照品/标准品1714528 长春瑞滨杂质A Vinorelbine Related Compound A 对照品/标准品1714506 酒石酸长春瑞滨 Vinorelbine Tartrate 对照品/标准品1714007 硫酸长春新碱 Vincristine Sulfate 对照品/标准品1713004 硫酸长春碱 Vinblastine Sulfate 对照品/标准品1711508 阿糖腺苷 Vidarabine 对照品/标准品1711472 维替泊芬杂质A Verteporfin Related Compound A 对照品/标准品1711461 维替泊芬 Verteporfin 对照品/标准品1711440 维拉帕米杂质F Verapamil Related Compound F 对照品/标准品1711439 维拉帕米杂质E Verapamil Related Compound E 对照品/标准品1711428 维拉帕米杂质D Verapamil Related Compound D 对照品/标准品1711406 维拉帕米杂质B Verapamil Related Compound B 对照品/标准品1711304 维拉帕米杂质A Verapamil Related Compound A 对照品/标准品1711224 维库溴铵杂质F Vecuronium Bromide Related Compound F 对照品/标准品1711202 盐酸维拉帕米 Verapamil Hydrochloride 对照品/标准品1711188 维库溴铵杂质C Vecuronium Bromide Related Compound C 对照品/标准品1711177 维库溴铵杂质B Vecuronium Bromide Related Compound B 对照品/标准品1711166 维库溴铵杂质A Vecuronium Bromide Related Compound A 对照品/标准品1711155 维库溴铵 Vecuronium Bromide 对照品/标准品1711133 赖氨加压素 Lypressin 对照品/标准品1711100 加压素 Vasopressin 对照品/标准品1711009 香草醛熔点标准品 Vanillin Melting Point Standard 对照品/标准品1710006 香草醛 Vanillin 对照品/标准品1709018 Vancomycin B with Monodechlorovancomycin 对照品/标准品1709007 盐酸万古霉素 Vancomycin Hydrochloride 对照品/标准品1708795 缬沙坦杂质 C Valsartan Related Compound C 对照品/标准品1708784 缬沙坦杂质 B Valsartan Related Compound B 对照品/标准品1708773 缬沙坦杂质 A Valsartan Related Compound A 对照品/标准品1708762 缬沙坦 Valsartan 对照品/标准品1708751 戊柔比星分离度用混合物 Valrubicin Resolution Mixture 对照品/标准品1708730 戊柔比星 Valrubicin 对照品/标准品1708729 丙戊酸杂质A Valproic Acid Related Compound A 对照品/标准品1708718 丙戊酸杂质 B Valproic Acid Related Compound B 对照品/标准品1708707 丙戊酸 Valproic Acid 对照品/标准品1708503 L- 缬氨酸 L-Valine 对照品/标准品1708015 D-缬更昔洛韦 D-Valganciclovir 对照品/标准品1708004 缬更昔洛韦盐酸盐 Valganciclovir Hydrochloride 对照品/标准品1707908 缬草烯酸 Valerenic Acid 对照品/标准品1707894 万乃洛韦杂质G Valacyclovir Related Compound G 对照品/标准品1707883 万乃洛韦杂质F Valacyclovir Related Compound F 对照品/标准品1707872 万乃洛韦杂质E Valacyclovir Related Compound E 对照品/标准品1707861 万乃洛韦杂质D Valacyclovir Related Compound D 对照品/标准品1707855 万乃洛韦杂质C Valacyclovir Related Compound C 对照品/标准品1707839 盐酸万乃洛韦 Valacyclovir Hydrochloride 对照品/标准品1707806 熊去氧胆酸 Ursodiol 对照品/标准品1706701 C13尿素 Urea C 13 对照品/标准品1706698 尿素 Urea 对照品/标准品1706009 乌拉莫司汀 Uracil Mustard 对照品/标准品1705800 阿糖尿苷 Uracil Arabinoside 对照品/标准品1705505 十一烯酸 Undecylenic Acid 对照品/标准品1705323 泛癸利酮杂质A Ubidecarenone Related Compound A 对照品/标准品1705312 系统适用性试验用泛癸利酮 Ubidecarenone for System Suitability 对照品/标准品1705301 泛癸利酮 Ubidecarenone 对照品/标准品1705006 L- 酪氨酸 L-Tyrosine 对照品/标准品1704003 泰洛沙泊 Tyloxapol 对照品/标准品1703850 酒石酸泰洛星 Tylosin Tartrate 对照品/标准品1703805 泰洛星 Tylosin 对照品/标准品1702008 氯筒箭毒碱 Tuboc
液相的方法上供试品和对照品的配液浓度不一样,一般我们是按照供试品还是对照品的浓度来配制进样浓度呢?
问:常用菌种及培养基英文对照答:Bacillus subtilis 枯草芽孢杆菌 Bacillus pumilus 短小芽胞杆菌 Staphylococcus aureus 金黄色葡萄球菌 Micrococcus luteus藤黄微球菌 Escherichia coli 大肠埃希杆菌 Saccharomyces cerevisiae啤酒酵母菌 Candida albicans 白色念珠菌 Aspergillus niger黑曲霉 Salmonella paratyphi B 乙型副伤寒沙门(氏)菌 Pseudomonas aeruginosa铜绿假单胞菌 Coliform 大肠菌群 Clostridium 梭菌 Nutrient agar 营养琼脂培养基 Rose Bengal agar Sodium 玫瑰红钠琼脂培养基 Nutrient broth medium营养肉汤培养基 Thioglycollate medium 硫乙醇酸盐流体培养基 Martin medium modified 改良马丁培养基 Bile salt lactose enrichment 胆盐乳糖培养基 0.1% peptone water medium 0.1% 蛋白胨水 pH 7.0 sodium chloride peptone buffer pH 7.0氯化钠-蛋白胨缓冲液 Lactose bile salt fermentation medium 乳糖胆盐发酵培养基[
请教各位前辈:1.听说有个ZN1009-2004标准,能检测水产品中,哪位前辈有,能否给我邮一份?2.有其他能同时前处理和检测水产品中恩诺沙星、环丙沙星、诺氟沙星、氧氟沙星的方法吗?QQ:3733818E-mail:yardin@126.com谢了先!!
[color=#444444]现在在测多巴胺注射液中的辅料EDTA,含量约50微克/毫升,网上查了用C18柱,四甲基氢氧化铵-水-乙腈作流动相,进对照品时峰形良好,浓度放大时峰面积也基本成线性增大,但是测供试品时,浓度在2微克峰就前沿了,放大浓度峰面积也不变,前沿更严重是为什么?供试品和对照品溶液配样时都加了铜离子络合[/color][color=#444444][color=#444444]文献里用的四丁基氢氧化铵,我暂用了四丁基溴化铵代替[/color][/color]
粪大肠菌群347.2 2018中阴性和阳性对照的意义是什么?验证的是培养基吗?为什么要做阴阳性对照实验?阴性对照的现象是什么呢?求解答
参考《中国药典》2010年版 取大肠埃希菌,金葡菌,铜绿菌,各50-100cfu,分别注入无菌平皿中,立即倾注胰酪胨大豆琼脂培养基,每株试验菌平行制备2个平皿,混匀。凝固,置30-35℃培养48h,计数。 取白念、黑曲各50-100cfu,分别注入无菌平皿中,立即倾注沙氏葡萄糖琼脂培养基,每株试验菌平行制备2个平皿,混匀,凝固,置20-25℃,培养72h,计数。 同时,用对应的对照培养基替代,被检查的培养基,进行上述试验。 结果判断: 1.若被检的培养基上的菌落平均数不小于对照培养基上的菌落平均数的70%;2.菌落形态大小与对照培养基上的菌落一致,则 判定该培养基的适用性符合规定。
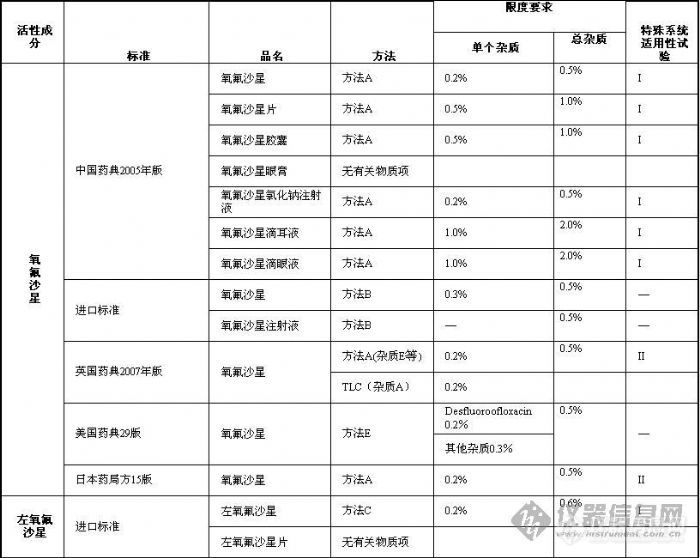
作者 张立雯 成海平 正文内容 【摘要】本文总结了国家标准中氧氟沙星、左氧氟沙星、盐酸左氧氟沙星、乳酸左氧氟沙星、甲磺酸左氧氟沙星系列药物的有关物质控制方法,分析了该类药物注册申报中有关物质控制存在的问题,希望能为研发者提供帮助。 【关键词】氧氟沙星、左氧氟沙星、有关物质一、概况 氧氟沙星(Ofloxacin)为合成的第三代广谱氟喹诺酮类抗菌药,对大多数革兰氏阳性菌和革兰氏阴性菌均有明显的抑制作用。临床上主要用于敏感菌所致的呼吸系统感染、泌尿生殖系统感染。氧氟沙星由日本第一制药株式会社研发,于1985年在日本、德国上市,制剂为口服片剂、注射剂等。目前国内已上市的氧氟沙星制剂有片剂、胶囊剂、颗粒剂、缓释制剂、小针、葡萄糖注射液和氯化钠注射液等。 左氧氟沙星(Levofloxacin)为氧氟沙星的左旋体,具有抗菌谱广、抗菌作用强的特点。日本第一制药株式会社于1993年在日本上市销售左氧氟沙星原料及片剂,并现已在英国、美国等多国上市。目前国内上市的左氧氟沙星制剂主要有片剂、小针、葡萄糖注射液和滴眼剂等。另外,国内已批准上市的左氧氟沙星还有其盐酸盐、乳酸盐和甲磺酸盐,三种加酸根的左氧氟沙星均有片剂、胶囊剂、注射制剂等多种剂型上市。二、国家标准中有关物质控制方法比较 氧氟沙星系列药物的有关物质测定国家标准大多采用HPLC法,列表比较见表1。 表1 氧氟沙星系列药物的有关物质测定方法与限度的比较 这些方法有很多相似的地方,如均采用ODS柱,色谱条件与含量测定色谱条件相同,按照主成分自身稀释对照法定量等。但也有一些不同的地方值得关注,作者从以下三个方面来对这些国家标准方法的不同之处进行比较。1、流动相 按照流动相的不同,作者将有关物质测定方法分为六种,具体如下: 方法A:醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH至 2.2)-乙腈(85∶15)为流动相,在294nm下检测; 方法B:略; 方法C:略; 方法D:己烷磺酸钠[取己烷磺酸钠0.98g,加磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水溶解并稀释至 1000ml,加0.05mol/L磷酸约500ml,使pH为2.4)]-甲醇(3∶1)为流动相,在293nm下检测; 方法E:磷酸缓冲溶液(溶解27.2磷酸二氢钾在1000ml水中,用磷酸调节pH至2.4)-乙腈(90∶10)为流动相,在294nm下检测; 方法F:己烷磺酸钠[取己烷磺酸钠0.98g,加磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水溶解并稀释至 1000ml,加0.05mol/L磷酸约500ml,使pH为2.4)]-甲醇(65∶35)为流动相,在230nm下检测。 六种流动相的共同特点是:组成均是酸性缓冲溶液加有机溶剂(甲醇或乙腈)。方法A、B、E未加表面活性剂,方法C、D加有表面活性剂己烷磺酸钠。除方法F在230nm下检测外,其他方法均在294或293nm下检测。2、主要杂质 英国药典收载了氧氟沙星杂质A、B、C、D、E、F共6个已知杂质,依次分别为去哌嗪环、去羧基、去氟、氟取代位置不同、去甲基以及氮氧化的化合物物。英国药典氧氟沙星原料药采用TLC法控制杂质A,采用HPLC法控制其他已知和未知杂质。美国药典重点关注了杂质desfluoroofloxacin,日本药局方重点关注了ofloxacin demethyl substance,均与英国药典的杂质E相同,是氧氟沙星的去甲基化合物,该化合物为氧氟沙星的主要降解产物,光照下极易产生。美国药典给出了desfluoroofloxacin的相应因子为1.13。中国药典没有明确已知杂质,但有关物质检查时采用以下光照降解法进行特殊系统适用性试验,光照试验中产生的杂质即为氧氟沙星、左氧氟沙星的主要杂质。 美国药典中desfluoroofloxacin按加校正因子的主成分自身稀释对照法定量,美国药典氧氟沙星其他杂质和其他国家标准中氧氟沙星或左氧氟沙星所有杂质按不加校正因子的主成分自身稀释对照法定量。有关物质限度的要求详见表1。3、特殊系统适用性试验 氧氟沙星、左氧氟沙星及其盐的含量测定和有关物质检查方法的系统适用性试验除通常的进样精密度、记录时间、理论塔板数等的要求外尚有一项较为特殊的系统适用性试验,其他标准方法采用I法,日本药局方采用II法,详述如下: 特殊系统适用性试验I(光照降解法):取供试品溶液于无色试管中,用日光灯(2500lux或3500lux)或紫外灯(254nm)照射1小时或3小时或4小时,取此液注入液相色谱仪,记录色谱图,相对保留时间约为主峰1.2处应能检测出色谱峰。 特殊系统适用性试验II(杂质对照品法):氧氟沙星和杂质E(ofloxacin impurity E CRS,英国药典)或氧氟沙星的去甲基物(ofloxacin demethyl substance)分离度不得低于2.0或2.5。 由于缺少杂质对照品,国内氧氟沙星系列药物的有关物质测定系统试验常常是采用光照降解法。也正是因为缺少杂质对照品,系统适用性试验才显得尤为重要,是考察系统分离能力的重要指标。 另外,左氧氟沙星及其盐的原料和制剂均需检查右旋异构体,方法基本相同,均是采用硫酸铜-L异亮氨酸溶液-甲醇或硫酸铜-D苯丙氨酸溶液-甲醇为手性流动相检测,限度要求不得过0.8%或1.0%。在此就不详加讨论。三、注册申报中存在问题与探讨1、不重视系统适用性试验 氧氟沙星系列药物的特殊系统适用性试验常常被忽视,其实却非常重要。若不做该项试验,就不能保证所采用的系统能将最难分离的相对保留时间1.2倍的色谱峰分离出来,就有可能得到错误的结果。 审评中曾发现申报盐酸左氧氟沙星注射液的某厂家自测盐酸左氧氟沙星含量较药检所检验结果高约5%(含量测定色谱条件与有关物质检查一致)。仔细审查其图谱,发现未按盐酸左氧氟沙星注射液的已有国家标准用光照降解法进行系统适用性试验,且色谱峰明显拖尾。其测定结果偏高很可能是紧随主峰之后的杂质峰包裹进了主峰。 申请人往往会留意进样精密度、理论塔板数这样的常规系统适用性试验,却常常忽略了光照降解系统适用性试验,此种现象在申报资料中占很大比例。究其原因,是试验人员没有理解到此项系统适用性试验的目的和重要性,希望提醒申请人提高对系统适用性试验的重视程度。2、没有杂质个数与含量的详细对比 申报资料中杂质对比研究通常的做法就是按照国家标准方法检验一下自制品和已上市对照药品,若都在标准规定范围内,就认为自制品与已上市药品质量相当。其实这样的做法是对杂质对比的目的和比什么不甚明了的表现。杂质对比一方面要了解自己的产品与已上市品杂质有哪些不同,另一方面要了解制剂过程中有没有新产生的杂质,若有新产生的杂质,应加以控制。所以比较就要落到列表对比杂质的个数与含量上,泛泛地比较杂质总量是不足以说明问题的。 例如,申报盐酸左氧氟沙星、乳酸左氧氟沙星的注射剂采用方法D测定的较为多见。在该色谱条件下,相对保留时间为0.23、0.43、1.2左右的杂质峰较常见,其中相对保留时间为1.2的色谱峰是稳定试验中含量有所增加的主要杂质。 另外,统计杂质的个数时要注意“忽略限度”。英国药典氧氟沙星有关物质项下明确规定:Disregard any peak with an area less than 0.1 times the area of the principal peak in the chromatogram obtained with reference solution (a),即忽略面积小于对照溶液主峰面积0.1倍的色谱峰(0.02%)。中国药典没有这么详细的规定,但从实际操作来看,为增强方法的严谨性和数据的可比性,建议申请人在统计杂质个数时应明确忽略限度。3、强力破坏试验降解程度不合适 有关物质检查方法学验证的重要项目就是通过强力破坏试验考察方法的专属性,但强力破坏试验破坏程度的掌握不尽合理。较容易出现的情况是破坏太轻微,酸、碱、氧化、光照破坏均几乎未产生可检测的杂质,这样就无法判断所采用的色谱条件分离能力是否符合要求,破坏试验失去意义。另一种极端是破坏过度,主峰降解了大半,产生大量重叠的杂质色谱峰,这样很难找到合适的色谱条件将所有的降解产物分离。个人认为,适度的破坏应是采用比贮藏中可能遇到的最强条件稍强烈的条件降解,产生比贮藏中可能产生的稍多杂质,若所选色谱条件能将这些杂质都分离,就是专属性符合要求的。 以上问题是氧氟沙星系列药物审评中经常遇到的问题,希望能为研发者提供帮助,共同努力提高我国仿制药的研发水平。 [img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907202227_160694_1612824_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907202231_160696_1612824_3.jpg[/img]
实验室采用3.4ml磷酸 三乙胺调pH=2.4可是这样的话,走混标工作液的时候,诺氟沙星和氧氟沙星是分不开的,怎么办?? 有谁知道?
莫西沙星混合对照品究竟有几个成分?5个还是包括莫西沙星在内的6个?Dissolve 5 mg of moxifloxacin for peak identification CRS (containing impurities A, B, C, D and E) in solution A and dilute to 5.0 ml with the same solution.
大家好: 我们的养殖场使用的氧氟沙星我们却在蛋中检测出诺氟沙星请大家分析一下原因,我们是用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测的。谢谢!
[size=4]1.所用对照品(标准品)中检所已经发放提供(可参阅中国药典2005年版二部附录ⅩⅤG),且使用方法相同时,应使用中检所提供的现行批号对照品(标准品),并提供其标签和使用说明书,说明其批号,不应使用其他来源者;如使用方法与说明书使用方法不同(如定性对照品用作定量用、效价测定用标准品用作理化测定法定量、UV法或容量法对照品用作色谱法定量等),应采用适当方法重新标定,并提供标定方法和数据;若色谱法含量测定用对照品用作UV法或容量法,定量用对照品用作定性等,则可直接应用,不必重新标定。 2.申报临床研究时,如中检所尚无供应,为不影响注册进度,可先期与中检所接洽制备和标定,申报时提供标定报告、标签(应标明效价或含量、批号、使用效期)和使用说明书;也可与省所合作标定,申报时提供标准品或对照品研究资料,“说明其来源、理化常数、纯度、含量及其测定方法和数据”;标定有困难时,可使用国外药品管理当局或药典委员会发放的对照品(标准品)或国外制药企业的工作对照品(标准品),进行标准制订和其他基础性研究,但应提供其标签(应标明其含量)和使用说明书,能保证其量值溯源性;也可使用国外试剂公司(如sigma公司等)提供的对照品(标准品),但应提供试剂公司该批对照品(标准品)的检测报告(用作含量测定时,应有确定的含量数据),如为高纯度试剂,提供了国外试剂公司检测报告(用作含量测定时,应有确定的含量数据)时,也可使用,并应能保证其量值溯源性,但申请人应及时与中检所接洽对照品(标准品)的标定事宜,临床研究期间完成此工作。 3.直接申报生产品种,如中检所尚无供应,可参照2中要求进行,并提供相应研究资料,但申请人在标准试行期间应与中检所接洽并完成的标定事宜。 4.对照品(标准品)标定的技术要求: 4.1.创新药物 应说明对照品(标准品)原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱),提供标定方法的研究和验证资料(如与原料药质量研究项下相同,可不再提供)、含量测定数据及经统计分析得到的对照品(标准品)含量结果,并说明进行临床前药学研究、药理毒理学研究所用样品的含量是否用该批对照品(标准品)确定或可用该批对照品(标准品)进行量值溯源。 ●纯度测定方法应选用色谱法,并采用两种以上不同分离机理或不同色谱条件并经验证的色谱方法相互验证比较,同时采用二极管阵列检测器或其它适宜方法检测HPLC法的色谱峰纯度,而后根据测定结果经统计分析确定对照品(标准品)原料的纯度。 ●对于组份单一、纯度较高的药物,对照品(标准品)标定方法宜首选可进行等当量换算、精密度高、操作简便快速的容量法。可根据药物分子中所具有的官能团及其化学性质,选用不同的容量分析方法,但应符合如下条件:(1)反应按一个方向进行完全;(2)反应迅速,必要时可通过加热或加入催化剂等方法提高反应速度;(3)共存物不得干扰主药反应,或能用适当方法消除;(4)确定等当点的方法要简单、灵敏;(5)标化滴定液所用基准物质易得,并符合纯度高、组成恒定且与化学式符合、性质稳定(标定时不发生副反应)等要求。 标定方法的选择要关注如下事项:(1)供试品的取用量应满足滴定精度的要求(消耗滴定液约20ml);(2)滴定终点的判断要明确,提供滴定曲线。如选用指示剂法,应考虑其变色敏锐,并用电位法校准其终点颜色。(3)为排除因加入其它试剂而混入杂质对测定结果的影响,或便于剩余滴定法的计算,可采用“将滴定的结果用空白试验校正”的办法;(4)要给出滴定度(采用四位有效数字)的推导过程。 标定结果要根据3个以上实验室各不少于15组测定结果经统计分析,去除离群值和可疑值后的结果,并报告可信限。 ●如该药物没有可进行等当量换算并符合要求的容量法时,可采用反复纯化的原料,色谱法确定纯度后扣除有关物质、炽灼残渣、水分和挥发溶剂等后的理论含量确定为标准品含量,以此为基准进行对照品(标准品)的换代和量值传递。 ●用于抗生素微生物检定法的第一代基准标准品可参照上述方法标定,如为多组份抗生素,其组份比例应与拟上市产品组份比例一致或接近,或以其中某一组份纯品为基准标准品,但要注意标准品换代时量值传递的恒定。 ●仅用于鉴别定性的化学对照品,注重其结构确证的研究资料,纯度和含量的要求一般可适当降低。 ●杂质对照品,用作限度要求时,应提供其来源(合成路线)、结构确证的研究资料,应具备较高的纯度和含量,并提供纯度和含量的的测定结果,提供质量控制标准。 4.2其他类别药物,可参照4.1要求进行。 ●用于抗生素微生物检定法的标准品须用上市国的国家标准品或原发厂的工作标准品为基准标准品进行标定。标定时采用的原料药应符合相应要求,并提供原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱)。标定须用现行版中国药典附录收载的“抗生素微生物检定法”-三剂量法,并提供详细的方法学研究,包括检定菌和培养基的选择、剂量和剂距选择、缓冲液选择(如与质量研究项下相同,可不再提供)。每次标定结果均应照“生物检定统计法-量反应平行线测定法(3.3)”法进行可靠性测验及效价计算。按照《药品注册管理办法》,上市药品质量标准所用标准物质均须由中检所负责标定和管理,药品研发过程中,研制单位应注意及时与中检所联系标定事宜,以保证研发工作的连续性。[/size]
请教牛奶中氟喹诺酮残留的样品前处理方法,是SPE提取,用的是lc/ms测定,主要是氧氟沙星,诺氟沙星,环丙沙星,恩诺沙星,洛美沙星!







 我要推广仪器
我要推广仪器
 下载APP
下载APP