为什么有些固体标准品/对照品是装在安瓿瓶,而有些是装在西林瓶中?二者有什么区别,装在安瓿瓶中的标准品/对照品是否开瓶即用完?不然如何保存
我在做氨苄西林钠聚合物,在以水为流动相B的时候,进样对照溶液,对照溶液严重拖尾,流速1.0。对照溶液浓度0.5mg/ml。在这个过程中调过流速0.8,但峰很宽;流速1.2只是出峰时间提前而已,拖尾问题没有改善。调过对照溶液浓度0.25mg/ml,拖尾仍然没有改善。水用的是注射用水,抽滤2遍。有关文献中又说对照溶液严重拖尾可以加0.5%葡萄糖溶液或0.01mol/l甘氨酸适量,抑制氨苄西林和葡聚糖凝胶的缔合。我两个都试过了,没有改善啊。这个适量真的是很难控制,几滴?几毫升?求求各位老师帮帮我吧,对照溶液严重拖尾啊!!!怎么办???
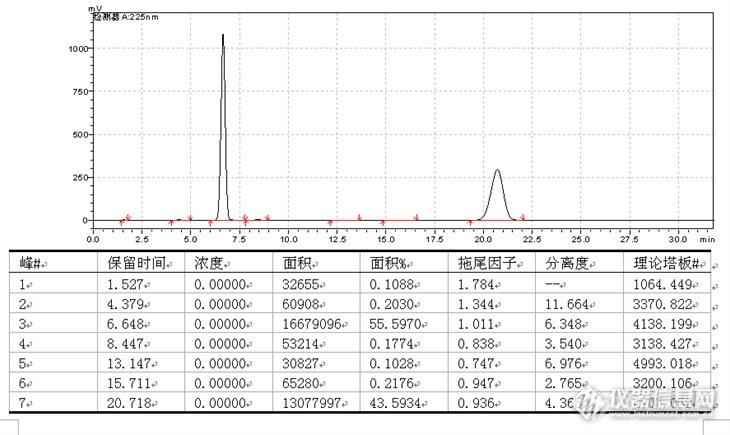
前言:这是一个老项目了,对于公开品名,是已经上报了,宝刀系列均是以前的资料,写出来和大家共享一下。由于网络问题,有些图看不到了,我在附件上显示了。建议大家看附件好了,看这个很吃力。望谅解。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(welchrom 填料:C18,规格:250mm×4.6mm,填料粒径:5μm;pn:wel518425,sn:w10212097)UV检测器(检测波长:225nm)柱温:室温流动相:流动相A为0.1mol/L磷酸二氢钾溶液-0.018mol/L十二烷基硫酸钠-甲醇-乙腈(275:275:200:250),用磷酸调节pH值至2.0;流动相B为乙腈。按下表进行线性梯度洗脱: 时间(分钟)流动相A(%)流动相B(%)090105851579010119010流速:1.2ml/min运行时间:约11分钟系统适用性:理论板数按阿莫西林峰和双氯西林峰计算应均不低于2000,双氯西林与阿莫西林的分离度应符合规定。具体试验操作:取装量差异项下的内容物,混合均匀,精密称取适量(约相当于阿莫西林25mg,双氯西林12.5mg),置100ml棕色量瓶中,加磷酸盐缓冲液(0.05mol/L磷酸二氢钾溶液-甲醇-乙腈(550:200:250)并稀释至刻度,摇匀,滤过,精密量取20μl注入液相色谱仪,记录色谱图;另取阿莫西林和双氯西林对照品,精密称定,加磷酸盐缓冲液(0.05mol/L磷酸二氢钾溶液-甲醇-乙腈(550:200:250)溶解并定量稀释制成每1ml中约含阿莫西林0.25mg和双氯西林0.125mg的溶液,同法测定,按外标法以峰面积分别计算出供试品中C16H19N3O5S和C19H17Cl2N3O5S的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。3.2.P.5.3.6 含量测定色谱图见附件1367~1442含量测定方法学验证结果概要 项目验证结果波长选择[size=9pt
[em09506]请问谁可以提供非洛地平氧化产物对照品?谢谢!

http://ng1.17img.cn/bbsfiles/images/2012/03/201203081038_353205_1638724_3.jpg上文中的阿莫西林系统适用性对照品就是含量测定用的对照品吗?
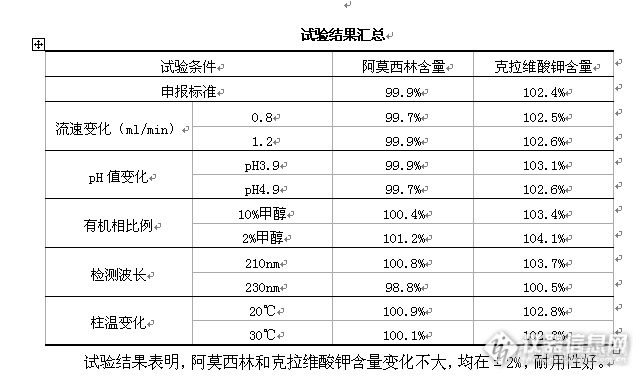
继”阿莫西林克拉维酸钾胶囊有关物质方法学”项目结束,整理的含量测定方法学。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-2010CHT (SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolution色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Xtimate C18 4.6*250 ,PN:Xt5B18425 ,SN:411101950UV检测器(检测波长:220nm)柱温:室温流动相:0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)。流速:1.0ml/min。运行时间:约20分钟。系统适用性:取阿莫西林克拉维酸系统适用性试验对照品,加流动相溶解并稀释制成每1ml中含0.8mg的溶液,取20μl注入液相色谱仪,记录的色谱图应与标准图谱一致。具体试验操作:取装量差异项下的内容物适量,精密称取适量,加水适量,超声使溶解并定量稀释制成每1ml中含阿莫西林0.5mg的溶液,滤过,立即精密量取续滤液20μl注入液相色谱仪,记录色谱图;另分别精密称取阿莫西林对照品与克拉维酸对照品各适量,加水溶解并定量稀释制成每1ml中约含阿莫西林0.5mg和每1ml中含克拉维酸0.125mg的混合溶液,同法测定。按外标法以峰面积分别计算供试品中C16H19N3O5S和C8H9NO5的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。“色”路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分http://ng1.17img.cn/bbsfiles/images/2013/06/201306292159_448382_1621890_3.gif3.2.P.5.3.6.1波长选择本品含量测定检测波长参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,即220nm。3.2.P.5.3.6.2流动相选择(色谱图见附件1122~1124)参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,以0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)为流动相。试验过程:系统适用性试验供试液:精密称取阿莫西林克拉维酸钾系统适用性对照品4.2mg至5ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;对照品溶液:精密称取阿莫西林对照品29.1mg和克拉维酸钾对照品7.3mg至50ml量瓶中,加水适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;精密量取上述供试液各20μl注入高效液相色谱仪,记录色谱图,典型色谱图见下图:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448383_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448384_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292201_448385_1621890_3.gif3.2.P.5.3.6.3进样精密度试验(色谱图见附件1125~1130)
求助氨苄西林红外光吸收图谱鉴别 标准规定:红外光吸收图谱应与对照的图谱一致。1、样品和对照怎么做前处理?
你购买过液相色谱系统适应性专用对照品吗,还是直接以含量用对照品来做,如阿莫西林?
有谁做过阿莫西林含量和有关物质,药典中提到色谱图应与标准图谱一致,标准图谱指的是什么,哪里有,含量中提到用系统适用性对照品,哪里可以买到?
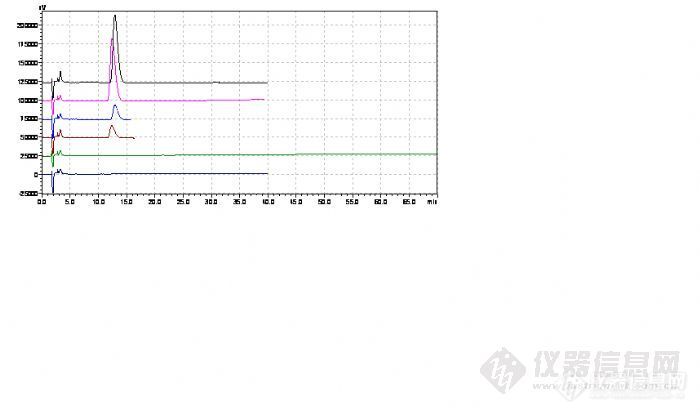
鉴于阿莫西林能引起人体过敏反应,目前在阿莫西林胶囊生产中产生的废水需进行阿莫西林灭活处理。处理的方式为废水与碱水(pH值大于13)反应1.5h。本次灭活验证分为3次灭活处理,每次分别在0.5h、1.0h、1.5h和2.0h取样检测阿莫西林。1.试验条件及仪器:仪器:LC-10AT VP(SHIMADZU CORPORATION)SPD-10A VP(SHIMADZU CORPORATION)工作站:LC solution(SHIMADZU CORPORATION)色谱柱:色谱柱信息:welchrom-C18,5μm,4.6*150mm; PN:wel518415,SN:W10211861流动相:0.05mol/L磷酸二氢钾溶液(用2mol/L氢氧化钾溶液调节pH值至5.0)—乙腈(97.5:2.5)检测波长:254nm,进样量:20μl,流速:1.0ml/min2.主要试剂及对照品:试剂:乙腈(HPLC级),磷酸二氢钾(AR),氢氧化钾(AR)阿莫西林对照品:中检所提供,批号:130409-200810,含量:86.9%3.试验过程:参照阿莫西林质量标准及检验操作规程(文件编号:RZ-SOP-QC04-001)、阿莫西林胶囊质量标准及检验操作规程(文件编号:RZ-SOP-QC06-001)进行检测废水处理后取样中阿莫西林的含量及中国药典2010年版二部收载的阿莫西林原料药和制剂胶囊质量标准。对照品溶液:取阿莫西林对照品适量加流动相稀释成每1ml约含阿莫西林1.0mg的溶液,滤过,取续滤液20μl注入高效液相色谱仪,记录色谱图。样品溶液:样液滤过,取续滤液20μl注入高效液相色谱仪,记录色谱图。10ppm-对照溶液:取本品(规格:0.25g,批号:20110601)16粒(含阿莫西林约4g,为本品阿莫西林胶囊说明书的最大日剂量)内容物,加流动相稀释制成每1ml约含阿莫西林0.04mg的溶液,滤过,取续滤液20μl注入高效液相色谱仪,记录色谱图。[/font
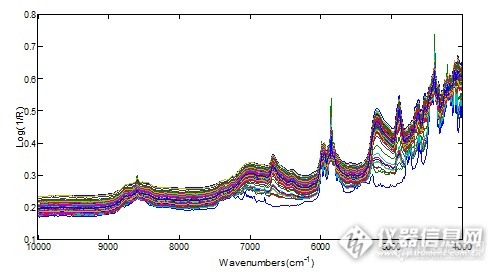
[align=center][b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术用于美洛西林钠舒巴坦钠药物混合过程在线混合均匀度终点监测[/b][/align][align=left][b]摘要: [/b]利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术,对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。[/align][b]关键词[/b]:[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url];分析模型;混合均匀度;在线监测自从2004年美国食品与药品监督管理局提出“过程分析技术”以来,全球的药品生产企业正在向着更高技术含量的生产方式和质量控制方式进军。近红外(Near infrared,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])光谱分析技术因其快速,无损的特点成为“过程分析技术”的重要组成部分,是制药企业进行产品中间体质量控制的重要方法之一。传统的检测方法为高效液相色谱法,紫外可见分光光度法等需要停止混合操作时才能取样检测,并且等待检测结果所需的时间也比较长,工作效率比较低,而[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱可以进行在线检测,连续记录不同混合时间内混合物的光谱图,建立数学模型对采集数据进行分析,从而判断各组分之间是否已经达到质量均一,工作效率大幅度的提高。本研究利用 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] 光谱分析技术在线监测美洛西林钠舒巴坦钠的药物混合过程,从而实现混合终点的准确判断。[b]1 材料1.1试剂[/b]美洛西林钠(13102041,山东瑞阳制药有限公司)舒巴坦钠(SS201310-26,江西东风制药有限公司)[b]1.2仪器和软件[/b]AntarisII型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国ThermoFisher公司),附有积分球采样模块;RESULT采样软件;电子分析天平(Sartorius BT224S,德国);TQ数据处理软件;表面皿;药匙;自制搅拌器。[b]2 方法2.1样品的准备[/b]精密称取舒巴坦钠固体原料药10.00g,美洛西林钠固体原料药40.00g,以备进行在线混合光谱的采集。平行制备3批样品,进行混合光谱的采集。[b]2.2模型的建立[/b]目前,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于混合过程在线监测的方法可分为活性药物成分(API)定量分析模型监测和基于移动块标准偏差(MBSD)的定性分析模型监测。前者为基于API药物含量的定量监测模型,当达到混合终点时,API的含量趋于一定值,可以依据模型监测的含量是否达到理论值并趋于稳定进行混合终点的监测;后者为基于光谱的标准偏差的定性监测模型。MBSD法的基本原理为:连续采集的若干张光谱间的标准偏差变化率趋于稳定并小于限定的一阈值时可认为达到了混合终点。其具体的计算步骤为:首先确定用于计算光谱标准偏差的光谱的条数n(即移动块的宽度),当[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析仪器采集到n张光谱后计算n张光谱的峰面积(或最大峰高、平均峰高等)的标准差,当采集到n+1张光谱时将第一张光谱移除,计算最近n张光谱的标准差,如此类推,最终得到随时间变化的光谱的标准偏差,根据标准差的变化进行混合终点的监测。本研究中建立了舒巴坦钠含量的定量分析模型和基于MBSD法的定性分析模型同时对用于混合终点的判断。[b]2.3在线混合光谱的采集[/b]将称取的美洛西林钠、舒巴坦钠原料药样品放入表面皿中,然后将表面皿放在Antaris II型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]积分球采样模块的上面,采用积分球漫反射采样方式进行光谱的采集。在运行在线混合工作流的同时采用自制的搅拌器进行样品的混合,采集得到混合过程的原始光谱,同时监测混合过程。波长范围10000-4000cm[sup]-1[/sup],每张光谱扫描次数4,混合过程中每间隔5s进行一张光谱的采集,光谱分辨率为8.0cm[sup]-1[/sup],每4个小时进行背景光谱的采集。每张[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱由1557个变量点组成。[b]2.4定量定性分析模型用于终点判断数据分析[/b]将在线混合过程进行监测,得到在线混合过程数据进行分析,以便了解混合全过程信息以及混合过程的监测。[b]2.5混合终点分析[/b]当得到混合终点时分别采集混合后的样品6处的原始[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,利用舒巴坦钠的定量分析模型预测混合终点时不同样品点处的舒巴坦钠的含量,判别是否混合均匀。[b]3 实验结果3.1分析模型的建立[/b]本研究中分别建立了在线混合过程的舒巴坦钠定量监测模型和基于移动块标准偏差的定性监测模型。[b]3.1.1 定性分析模型的建立[/b]目前混合均匀度在线监测常用的方法为MBSD法,本研究中MBSD法定性建模的参数为:选择的3个光谱区间包括全光谱、5275.6-4806.3cm[sup]-1[/sup](称为Region1)及7096.76-6344.66cm[sup]-1[/sup](称为Region2);用于计算光谱偏差的光谱的条数为5(即移动块的宽度为5)。[b]3.1.2 定量分析模型的建立[/b]本研究中所建立的定量分析模型用于监测混合过程中舒巴坦钠的百分含量的变化,因为本实验中舒巴坦钠和美洛西林钠两者间的混合比为4:1,当达到混合终点时,舒巴坦钠的百分含量应该在20%左右。其模型的具体参数见上一章中得到的舒巴坦钠百分含量的定量分析模型。[b]3.2混合在线过程数据分析[/b]本研究中平行进行了3次混合过程的在线监测,分别对3次实验结果进行分析,以充分了解混合监测过程。[b]3.2.1 第一批实验结果分析3.2.1.1 原始光谱图[/b]图1给出了混合过程中采集得到的208张原始光谱,由图中可知,处于下面的光谱较稀疏,可能属于混合刚开始的阶段,光谱会有较大的差异;处于上面的光谱较密集,其原因为随着混合的不断进行,光谱间差异越来越小,所以光谱较集中。[align=center][img=,498,274]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141912_01_1626619_3.png[/img][/align][align=center]图1 第一批混合过程原始光谱[/align][align=center] [/align][b]3.2.1.2 在线混合过程结果分析[/b]图2为定性分析模型中得到的3个光谱区间的峰面图,其中M1为全光谱建模的峰面积变化,M2为Region 1(5275.6-4806.3cm-1)的峰面积变化,M2为Region 2(7096.76-6344.66cm-1)的峰面积变化,由峰面积的变化图可知,混合过程的前100s其变化较为明显,M1不断升高,M2和M3(7096.76-6344.66cm-1)不断下降,之后峰面积值趋于稳定。[align=center][img=,525,234]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141913_01_1626619_3.png[/img][/align][align=center]图2 光谱区间峰面积图[/align]图3为舒巴坦钠含量及标准偏差变化图,由图中显示在混合的初期阶段,尤其是前100s左右,四个表征混合均匀度的参数均有着较大的变化趋势,在200-300s间四个参数有稍微较小的波动,此后随着混合过程的不断进行,表征混合均匀度的四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右,舒巴坦钠和美洛西林钠混合较为均匀,达到了混合终点。由图可知前100s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,538,292]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141914_01_1626619_3.png[/img][/align][align=center]图 3 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align][align=left] 当达到混合终点时分别采集表面皿下6个点的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,根据建立的模型测定其舒巴坦钠的百分含量,看混合是否均匀。表2给出了用所建模型得到的6个点的舒巴坦钠的百分含量值,6个点舒巴坦钠的百分含量值在20%左右,说明混合较为均一,但是最大的值达到了22.41%,可能是由于混合装置过于简陋,加上是人为搅拌进行混合,不能达到很好的混合,部分地方没有进行很好的混合。从实验的可行性方面,初步证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]技术用于美洛西林钠舒巴坦钠混合的可行性。[/align][align=center]表1混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,570,70]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_01_1626619_3.png[/img][/align][b]3.2.2 第二批实验结果分析3.2.2.1 原始光谱图[/b]图4给出了第二批混合过程中采集得到的203张原始光谱,其混合过程原始光谱的特征和第一批混合过程较为相似,混合初期光谱变化较为明显,随着混合的进行,光谱差异变小,光谱较为密集。[align=center][img=,488,280]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_02_1626619_3.png[/img][/align][align=center]图4 第二批混合过程原始光谱[/align][align=left] [b]3.2.2.2 在线混合过程结果分析[/b][/align]图5为各个光谱波段峰面积的变化图,由图中显示开始的100s内峰面积有着较大的变化幅度,随着混合的不断进行,峰面积的变化趋势不断减小并逐渐趋于稳定。[align=center][img=,516,307]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141916_01_1626619_3.png[/img][/align][align=center]图5 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图6为舒巴坦钠含量及标准偏差变化图,由图可知在混合的初期阶段大约0-100 s时,舒巴坦钠百分含量值及峰面积的标准偏差值有着明显的变化,全光谱峰面积的标准偏差(Full Range STD)在200-400 s间有较为明显的波段,此后随着混合过程的不断进行,四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右。由此可知前100 s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,551,327]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141917_01_1626619_3.png[/img][/align][align=center]图6 含量和标准偏差变化图[/align][align=center](a 舒巴坦钠百分含量 b 全光谱峰面积标准偏差 c Region 1峰面积标准偏差 d Region 2峰面积标准偏差)[/align]当达到混合终点时,采集表面皿底部6处的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,检测混合过程是否达到均一,表2列出来了6处的舒巴坦钠的百分含量值,由表2可知达到混合结束后得到的6处的舒巴坦钠的百分含量均在20%左右,说明混合较为均匀。同时,由于实验条件的限制加上搅拌时人为因素的影响等,各点之间含量也着较大的差异。[align=center]表2 舒巴坦钠百分含量[/align][align=center] [img=,566,84]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141918_01_1626619_3.png[/img][/align][b]3.2.3 第三批实验结果分析3.2.3.1 原始光谱图[/b]图7给出了混合过程中采集得到的207张原始光谱,由图中可知,得到的原始光谱图与第一批和第二批有着相似的结果,即混合的初期光谱差异大,因此光谱较为稀疏(偏下方的光谱),随着混合的进行,光谱间差异变小,光谱变得密集(偏上方的光谱)。[align=center][img=,505,262]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_01_1626619_3.png[/img][/align][align=center]图7 第三批混合过程原始光谱[/align][b]3.2.3.2 在线混合过程结果分析[/b]图8给出了混合过程中3个光谱区间峰面积的变化趋势值,由图中可知0-100s间三个光谱区间的峰面积有着明显的变化,100-200s间峰面积有着明显的变化,但是变化幅度没有前100s大,200s以后峰面积变化趋势变小。说明前200s是混合的主要阶段,峰面积变化较为明显。[align=center][img=,519,343]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_02_1626619_3.png[/img][/align][align=center]图 8 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图9为舒巴坦钠百分含量及光谱峰面积的标准偏差随时间变化的趋势图,其变化趋势和峰面积的变化趋势相似,前100s变化幅度较大,100-200s间也有较为明显的变化,但是变化幅度不是很明显,200s后舒巴坦钠的百分含量和峰面积的标准偏差均趋于稳定,说明此时光谱差异变小,混合趋于均匀。[align=center][img=,529,352]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141920_01_1626619_3.png[/img][/align][align=center]图9 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align]表3为达到混合终点时采集表面皿底部的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱得到的不同点的舒巴坦钠的百分含量值,由表中显示6个点的舒巴坦钠的百分含量值在20%左右,但是6个点之间舒巴坦钠百分含量间存在较大的差异,测得的最小值为17.80%,其原因可能是一方面由于实验条件的限制混合不够均匀,一方面用于舒巴坦钠含量测定的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]定量分析模型也有一定的偏差,可能引起含量检测的差异存在。[align=center]表3 混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,564,66]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141921_01_1626619_3.png[/img][/align][b]3.3小结[/b]通过3个混合平行实验的进行可知所建立的基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型能够有效的监测舒巴坦钠、美洛西林钠的混合过程。由舒巴坦钠百分含量和标准偏差变化图可知两者的变化有着相关性,当舒巴坦钠的百分含量变化幅度大时,其标准偏差的变化幅度也较大,因此两者均可以用于混合过程的在线监测,证实了实验的可行性。[b]4 结论和讨论[/b]本研究采用AntarisII傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,然后Antaris II傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]漫反射采样方式采集混合过程中的光谱,实时监测混合过程的进行。通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。此外,MBSD法因为无需进行一级数据的采集,方法较为简单且容易理解,目前常用于混合过程的在线监测。本研究中有效证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术在舒巴坦钠美洛西林钠样品在线混合过程中应用的可行性,在样品的在线混合监测中有着重要的应用价值和应用前景。该技术能够克服传统方法费时、繁琐等缺点,而且可以实现过程的实时在线监测,让生产者充分了解整个生产过程中的参数变化。 [b]参考文献[/b]陆婉珍, 褚小立. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]([url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])和过程分析技术(PAT). 现代科学仪器, 2007(004):13-17.SieslerH, Ozaki Y, Kawata S, et al. Near-infrared spectroscopy: principles .Instruments, Applications, 2002:35-181.Bhushan,K.R.,et al.Detection of breastcancer microcalcifications using a dual-modality SPECT/[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] fluorescent probe. J Am Chem Soc, 2008. 130(52):17648-17649.贾燕花. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术在化学药品生产过程控制应用初探. 北京协和医学院, 2011.Fevotte.G,et al.Applications of [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]spectroscopy to monitoring and analyzing the solid state during industrialcrystallization processes . Int J Pharm, 2004, 273(1):159-169.张敏.盐酸林可霉素多晶型分子构象对其红外光谱行为的影响.中国抗生素杂志, 2005, 30(009):529-532.Blanco M,R Goz"01ez Ba,E.Bertran,Monitoring powder blending in pharmaceutical processes by use of nearinfrared spectroscopy . Talanta, 2002, 56(1):203-212,田科雄.不同装载系数和混合时间对添加剂预混料混合均匀度的影响.河北畜牧兽医, 2004, 20(9):52-53.孙栋. 基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的几种固体粉末混合均匀度快速检测研究. 山东大学硕士学位论文, 2012年.
102%(对照为奥美拉唑钠),后来领导指示我们用奥美拉唑对照再来用液相检测奥美拉唑钠,含量又和奥美拉唑钠对照算出来的结果差很多(奥美拉唑钠和奥美拉唑的转换系数考虑在内了),大家帮忙分析一下是什么原因啊
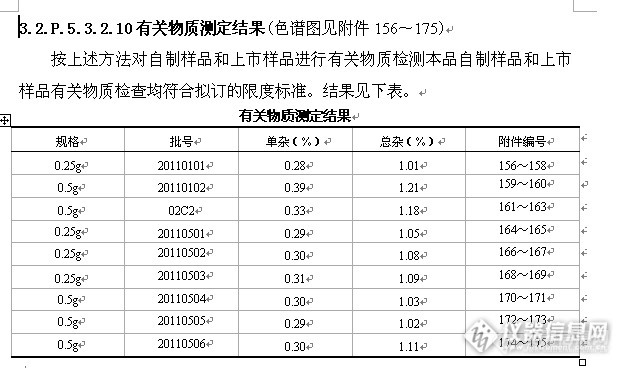
继“宝刀未老,刀走偏锋,阿莫西林舒巴坦匹酯片含量测定方法学部分”,根据该项目整理后,于是“接步献刀”进行的有关物质方法学研究,本品国内外药典均无收载且是复方制剂,根据相关的转正标准进行试验,均不理想,对有关物质的研究有一定的难度,根据相关资料进行研究,下文主要简介比较重要的部分如流动相的摸索和耐用性试验以及波长的选定,其他的同前几篇,具体如下:项目:有关物质(3.2.P.5.2.4)检查方法:HPLC法试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Wel5184425,sn:w10212096,ln:w1801.19UV检测器(210nm)柱温:室温流动相:0.01mol/L十二烷基硫酸钠溶液(用磷酸调节pH值至2.5±0.02)-甲醇(45:55)为流动相流速:1.0ml/min运行时间:约55分钟系统适用性:理论板数按阿莫西林峰计算不低于2000。具体试验操作:取本品细粉适量,精密称定,用流动相适量溶解并稀释制成每1ml中约含阿莫西林和舒巴坦均为0.5mg的溶液,摇匀,滤过,取续滤液作为供试品溶液;精密量取供试品溶液1ml置100ml量瓶中,用流动相稀释至刻度,作为对照溶液。取对照溶液10μl注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的20%~25%。再精密量取供试品溶液和对照溶液各10μl,分别注入液相色谱仪,记录色谱图,对照溶液中阿莫西林的峰面积As,供试品溶液中各杂质的峰面积Ai均通过自动积分法测定,以各杂质峰面积与对照溶液阿莫西林峰面积的比值计算得出各杂质的含量,总杂质为各杂质的和。计算公式:各杂质的量(%)=Ai/As杂质总量(%)=∑ihttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300029_448425_1621890_3.png3.2.P.5.3.2.1 流动相选择该品种目前在中国药典、美国药典、日抗基和英国药典均未收载。参照新药转正标准第55册收载的阿莫西林舒巴坦匹酯片质量标准WS1-(X-145)-2004Z、进口药品注册标准JX20080076、舒巴坦匹酯国家质量标准WS-516(X-439)-2001(试行)以及阿莫西林舒巴坦匹酯制剂的其他相关研究资料(傅小雅.HPLC-DAD法测定阿莫西林舒巴坦匹酯分散片中舒巴坦匹酯含量及有关物质.中国药房2008年第19卷第13期:1011-1012;姜红,胡昌勤,金少鸿.阿莫西林-舒巴坦匹酯片含量及有关物质质控分析方法的建立.药物分析杂志2002,22(2):91-94)进行有关物质检查流动相选择。由于阿莫西林和舒巴坦匹酯极性差别较大,通常会造成阿莫西林在死时间附近出峰,而舒巴坦匹酯出峰时间较迟。本品试验表明流动相加入十二烷基硫酸钠溶液能有效检查有关物质。初步拟定流动相体系及试验结果统计见下表。http://ng1.17img.cn/bbsfiles/images/2013/06/201306300030_448426_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300031_448427_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300031_448428_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300032_448429_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300033_448430_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300033_448431_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300034_448432_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300035_448433_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300035_448434_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300036_448435_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300037_448436_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306300038_448437_1621890_3.png3.2.P.5.3.2.4有关物质检查波长选定(色谱图见附件115~117)本品目前在中国药典、美国药典、日抗基和英国药典均未收载。参照新药转正标准第55册收载的阿莫西林舒巴坦匹酯片质量标准WS
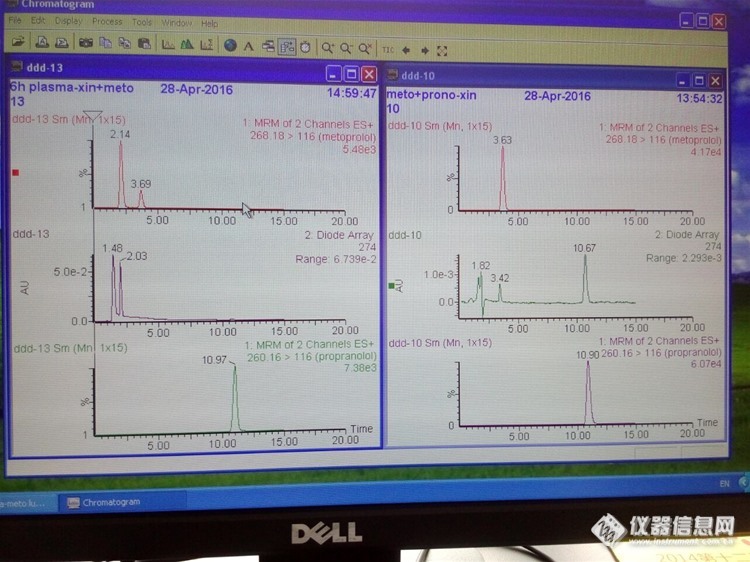
麻烦各位朋友帮忙分析一下了,困扰了我很久了,很多方法都试了,还是那样。情况介绍:流动相为甲醇-乙腈-0.2%甲酸水=10:15:75;样品为琥珀酸美托洛尔灌胃大鼠后血浆样品,图谱ddd-13;样品和对照品均用流动相作溶剂,对照品浓度远大于样品,图谱ddd-10;进样量5ul;MRM扫描。主要问题:质谱图提取后,对照品中美托洛尔是单峰;样品中美托洛尔是双峰,第一个峰的峰面积远大于第二个峰。样品为何会出双峰,真的都是美托洛尔?为何色谱保留时间差别很大?http://ng1.17img.cn/bbsfiles/images/2016/04/201604291443_591954_3099769_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/04/201604291444_591955_3099769_3.jpgfile:///C:\Program Files\Tencent\QQ\Users\540761932\Image\C2C\C8E4ADEAE357ABD265B94368610BE8C5.jpg

因本品按照以前的转正标准,无法达到分析要求,于是刀走偏锋,另辟蹊径,以下是本品含量测定方法学研究内容:项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-10AT VP(SHIMADZU) SPD-10A VP(SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolutionlite色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Wel5184425,sn:w10212096,ln:w1801.19UV检测器(210nm)柱温:室温流动相:磷酸二氢钾溶液(取磷酸二氢钾3.06g,加水900ml溶解后,用磷酸调节pH值至3.0,再加水稀释至1000ml,混匀)-乙腈(40:60)。流速:1.0ml/min运行时间:约10分钟具体试验操作:取本品20片,精密称定,研细,精密称取细粉适量(约相当于阿莫西林和舒巴坦分别为50mg),置100ml量瓶中,加流动相适量,超声使阿莫西林和舒巴坦匹酯溶解,加流动相稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;另分别取阿莫西林和舒巴坦匹酯对照品适量,加流动相制成每1ml中分别含0.5mg阿莫西林和舒巴坦的溶液,作为对照品溶液。精密量取供试品溶液和对照品溶液各10μl,分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算,即得。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液中相应主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液中相应主峰面积;W为供试品取样量(mg)。http://ng1.17img.cn/bbsfiles/images/2013/06/201306292348_448407_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292348_448408_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292349_448409_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292350_448410_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292350_448411_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292351_448412_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292351_448413_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292352_448414_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292353_448415_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292353_448416_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292354_448417_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292354_448418_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292355_448419_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292356_448420_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292357_448421_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292358_448422_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292358_448423_1621890_3.pnghttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292359_448424_1621890_3.png
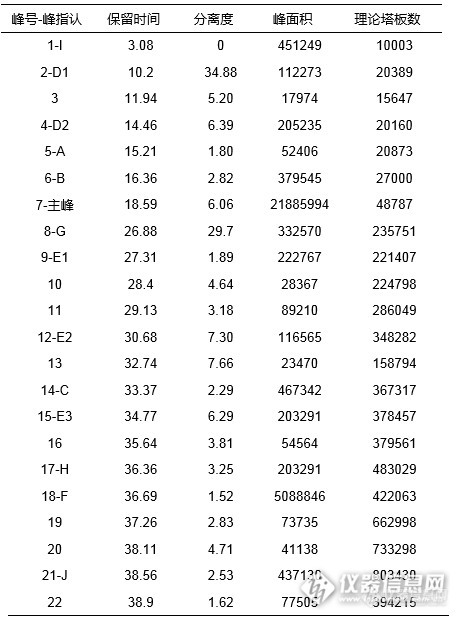
[align=center][b][color=black]阿莫西林及其杂质的液相分析[/color][/b][/align][b][/b][align=left]首先参考客户提供液相条件,对客户提供的阿莫西林样品进行分析尝试。由于梯度条件水相较高,故使用可在100%水相条件下稳定使用的资生堂高极性C[sub]18[/sub]色谱柱CAPCELL PAK AQ;同时,为提高杂质间分离度,我们选择柱效更高的3μm粒径色谱柱进行实验条件的优化。实验中发现,柱温对阿莫西林杂质B与杂质D2,杂质G与杂质E1,以及杂质H与杂质F间的分离有较大影响,结果如图1所示。[/align][align=center][img=,690,305]http://ng1.17img.cn/bbsfiles/images/2017/06/201706150846_02_2222981_3.png[/img][/align][align=center][/align][align=center]图1 不同温度下分离情况比较[/align][align=left][b]HPLC Conditions[/b]色谱柱:CAPCELL PAK C[sub]18[/sub] AQ S3 4.6mm i.d.×250mm流动相:A : 磷酸缓冲盐* B : 磷酸缓冲盐*/ 乙腈= 80/ 20 25°C B% 0%(0min)→0%(10min)→100%(40min)→100%(45min)→0%(45.1min)→0%(55min) 30°C B% 0%(0min)→0%(10min)→80%(40min)→80%(45min)→0%(45.1min)→0%(55min) 35°C B% 0%(0min)→0%(10min)→70%(40min)→70%(45min)→0%(45.1min)→0%(55min)流 速:1.0 mL/ min温 度:25°C、30°C、35°C检 测:PDA 254nm浓 度:客户提供进样量:20µ L*注:磷酸缓冲盐:0.05mol/L 磷酸二氢钾,用2mol/L 氢氧化钾调节pH为5.0[/align][align=left]由图1,温度越高,杂质D2保留时间越短,在35℃时杂质D2与杂质A、杂质B得到了完全分离;杂质G和杂质E1在35℃时亦得到良好分离;而杂质H与杂质F在35℃条件下反而重合在一起,无法分离。经过多番考量,最终选择在柱温31℃条件下对梯度条件进行优化,得到图2结果,同时通过图3单标定位和客户参考谱图共同参考,标定杂质峰序(因有杂质出多个峰,指定仅供参考)。[/align][align=center][img=,690,390]http://ng1.17img.cn/bbsfiles/images/2017/06/201706150846_03_2222981_3.png[/img][/align][align=center]图2 阿莫西林对照品及放大谱图[/align][align=center](图中显示数字为分离度)[/align][align=center][img=,690,358]http://ng1.17img.cn/bbsfiles/images/2017/06/201706150846_04_2222981_3.png[/img][/align][align=center]图3 阿莫西林对照品及单标谱图[/align][align=center][img=,460,620]http://ng1.17img.cn/bbsfiles/images/2017/06/201706150846_01_2222981_3.png[/img][/align][align=center]表1 阿莫西林对照品峰表[/align][align=center][b][/b][/align][align=left][b]HPLC Conditions[/b]色谱柱:CAPCELL PAK C18 AQ S3 4.6mm i.d.×250mm流动相:A : 磷酸缓冲盐* B : 磷酸缓冲盐*:乙腈 = 8 : 2 B% 0%(0min)→0%(10min)→50%(30min)→100%(40min)→0%(40.1min)→0%(50min)流 速:1.0 mL / min温 度:31°C检 测:PDA 254nm浓 度:客户提供进样量:20µ L*注:磷酸缓冲盐:0.05mol/L 磷酸二氢钾,用2mol/L氢氧化钾调节pH为5.0[/align][align=center][/align]
东南科仪分享:阿莫西林为抗细菌口服抗生素制剂,应检查霉菌每1g中不得超过100个,因其对铜绿假单胞菌无效,还应检查铜绿假单胞菌一、使用设备 日本ALPCL-32L高压灭菌器(东南科仪提供),德国binder KBF恒温恒湿箱(东南科仪提供), 离心机(kokusan提供),意大利VELP 均质器(东南科仪提供)二、霉菌的检查 配制供试液:称取供试液10g。置0.9%无菌氯化钠溶液100ml中,用意大利VELP 均质器或其它方法进行混匀,作为供试液。三、培养基使用前处理采用日本ALPCL-32L高压灭菌器直接对琼脂培养基,使用器皿的高压灭菌。四、预处理:可采用以下方法1.稀释法:将供试液注入较大的培养集中,使该供试液稀释至不具抑菌作用的浓度。2.离心沉淀集菌法:将规定量的供试液,离心(3000r/min)30min,留底部集菌液约2ml,再稀释成原规定量的的供试液,如有不溶性药渣,可离心(500r/min)5min,取全部上层液,再集菌处理。3.薄膜过滤法:取定量供试液至稀释剂100mI中,摇匀,以无菌操作加入装有直径约50mm,孔径不大于0.15um±0.02um微孔滤膜的过滤器内,减压抽干后,用稀释剂冲洗滤膜3次,每次50-100ml,取出滤膜备捡。4.利用平皿菌落计数法,将供试液涂布在玫瑰红钠培养基上,在适宜的温度下进行培养,观察肉眼可见的霉菌菌落数。五、铜绿假单胞菌(绿脓杆菌)的检查检验程序:铜绿假单胞菌、分离、纯培养革兰氏染色镜检,及生化试验等步骤进行检查。检查方祛:取胆盐乳糖培养基3份,每份100ml,两份分别加入规定量的供试液,其中一份入对照菌液作为阳性对照,第3份加入与供试液等量的稀释液作为阴性对照。培养18-24h阴性对照应无菌生长,其余2份培养液划线接种于溴化十六烷基三甲铵琼脂培养基平板上培养18-24h, ;当阳性对照的平扳呈阳性菌落时,供试品的平板无菌落或无疑似菌落的生长,可判未检出铜绿假单胞菌。铜绿假单胞菌典型菌落呈扁平,无定形,周边扩散,表而湿润,灰白色,周围时有蓝绿色扩散。如生长菌落具有上述特征或疑似者,应挑选2-3个菌落,分别接种于营养琼脂养斜面上,培养18-24小时。取培养物做革兰染色,并作氧化酶试验。如革兰阴性杆菌、氧气酶试验阳性,及绿脓菌素试验阳性,可判检出铜绿假单胞菌。绿脓菌素阴性的培养物,应继续做硝酸盐还原产气实验、42℃生长实验、明胶液化实验均为阳性时,应判检出铜绿假单胞菌。附:日本ALPCL-32L高压灭菌器特点1、CL-32L 高压灭菌器的显示屏、键盘及压力表在同一个斜面上,令操作和读数都同样方便。键盘功能指示明确,并可保留预设模式,一键start即可自动完成高压灭菌器的全部工作2、快速锁盖杆可轻松地锁紧高压灭菌器的灭菌舱盖,自动铰链使拾起各放下舱盖都毫不费力。3、双内锁装置确保高压灭菌器操作过程中舱盖的锁紧,并使温度保持安全范围内。4、高压灭菌器的状态图像显示,可清晰显示当前的工作状态。5、定时空气排出,确保高压灭菌器内纯蒸汽的灭菌环境。6、可调自动蒸气排出,使高压灭菌器灭菌完后蒸汽慢慢地自动排出。7、ALP生产的高压灭菌器专为处理培养基设计的保温及融化功能。8、定时启动功能,可使高压灭菌器的灭菌工作按所需日期时间启动,使您随时可以开始工作。9、记忆功能,高压灭菌器的使用工作程序即使在断电后也不丢失,重新接上电源,程序即可恢复。10、多选功能,ALP CL-32L高压灭菌器满足各种工作需要:①热空气或真空干燥 ②样品温度探头使灭菌更为可靠 ③自动给水④预热功能可缩短升温时间,⑤冷却风机,缩短冷却时间⑥打印机 ⑦记录仪 11、三种标准工作模式, CL-32L高压灭菌器组合加热、灭菌、排气、保温、融合等各步骤,适应液体、固体、废料、培养基等不同样品的处理需求。增配干燥设置,即可提供带干燥的操作模式
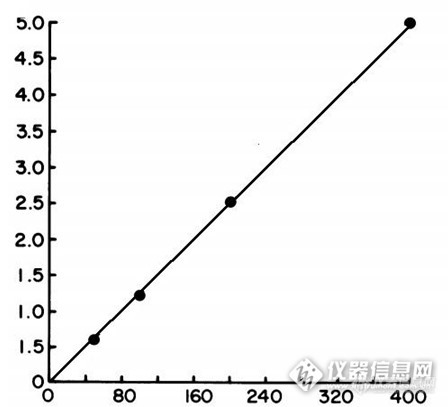
高效液相色谱法测定测定血清中替卡西林水平 卡替西林是一种半合成的抗假单胞菌青霉素,对于严重革兰阴性菌感染特别有效,除了用于治疗单胞菌感染,替卡西林也用于经验用于免疫受损的宿主。通常,这两种情况下,卡替西林总是与氨基糖苷类或头孢菌素联合应用。与青霉素联用的毒性一般是最小的,但当血清中水平高时,也会出现中枢神经系统的副作用。虽然不是常规要求,但是对于肾功能不全患者,特别与其他的β-内酰胺类抗生素联合用药时血清水平监测是很有必要的。 传统替卡西林的测定是通过微生物分析方法测定,该方法虽然划算,但这些方法总是缺乏与生化测定或免疫测定联用的特异性和精确度,而且需要最少8小时的孵育过程,不利于剂量调整。高效液相色谱法定量测定血清中替卡西林水平以及药剂中青霉素和头孢菌素和血清及尿液中替卡西林的测定在文献中均有报道,但均对于临床应用不宜,本实验做了调整优化,对于临床应用实用性较强。材料和方法: 替卡西林/替莫西林均购自药店,甲醇,氯仿,冰乙酸,盐酸,正戊醇,醋酸铵,磷酸二氢钠为分析纯或色谱纯。流动相为85(醋酸铵液):15(甲醇)醋酸铵液:醋酸铵液浓度为0.1M,并以冰醋酸调节PH为4 样品提取溶液预先配置室温保存:包含0.4N的盐酸,氯仿:正戊醇(3:1),0.1M磷酸盐缓冲液(PH=7),磷酸盐缓冲液用前按1:10用水稀释,去离子水。 标准,对照的配置:替卡西林二钠用灭菌的去离子水溶解后加入加热灭活的人血清中,配置浓度为50,100,200,400ug/ml,,并以同样方法配置250ug/ml作为对照。标准和对照血清分别以0.5ml分装,-70度保存。替莫西林以去离子水溶解于灭菌去离子水制成150ug/ml.,同法保存。标准和对照血清以及内标替莫西林用前融化。 样品制备:血清样品,标准和对照血清分别为0.35ml,加入0.15ml内标溶液,0.25ml 0.4N的盐酸,3.5ml的氯仿-正戊醇于带有螺旋盖的试管中。混合均匀后离心10分钟。上层弃去留下层。下层再加入0.35ml的磷酸盐缓冲液,混合均匀后离心10分钟。移取上层,4度保存备用。 液相条件:沃特斯2487配DAD检测器 water bondapak C18柱 (10um×4.6mm×150mm),检测波长242nm. 进样量20ml, 流速1.5ml/min 定量:标准曲线通过替卡西林的峰高与内标峰高的比率以及内标峰浓度进行绘制。 提取效率:替卡西林和内标的回收率通过比较血清提取以及相同浓素的含水制剂的峰高 精密度:日内通过向正常血清中加入替卡西林,(75ug/ml,150 ,ug/ml,300ug/ml),进行测定,日间通过三周内10次测定获得。 样品获得:该试验中应用的血清样本来自临床上那些替卡西林水平需要监测的患者。结果:1、血清中内标和替卡西林的提取后分析图谱如下:(内标和替卡西林的保留时间分别为5.4min,6.8min。http://ng1.17img.cn/bbsfiles/images/2014/10/201410300942_520789_2204138_3.png2、绝对回收率替卡西林血清回收率在29-385ug/ml范围内平均值为71%,而内标的回收率为67%,相对回收率,替卡西林在75-300ug/ml范围内平均为97%,如下图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410300943_520790_2204138_3.png3、下图为替卡西林标准曲线http://ng1.17img.cn/bbsfiles/images/2014/10/201410300943_520791_2204138_3.png讨论: 1、本实验开发了一种运用高效液相测定血液中替卡西林水平的方法,将血清加入替卡西林作为内表。采用氯仿-正戊醇进行萃取,后反萃取于磷酸盐缓冲液中。以反向C18柱,乙酸铵-甲醇水为流动相,240nm下进行检测。虽然头孢西丁,头孢噻吩,头孢呋辛等与替卡西林保留行为相似,但抗生素联合使用对于替卡西林的检测没有影响。试验表明本方法对于单用及联用抗生素时对于卡替西林的快速检测是准确,可重现的 2、本试验所采用的高效液相法分析血清中替卡西林的方法准确、重现性好,当患者联合用药时也能快速检测不干扰。 3、本试验采用内标的方法,从而克服了样品到样品间提取的变数,因为结构相似我们采用替莫西林作为内标。在提取过程和色谱行为方面也证明了采用替莫西林的可靠性。 4.该方法可用于抗生素联合用药时患者血清中替卡西林的水平测定,在患者的服用剂量调整范围内也是可适用的。
[color=#333333]对照品与标准品概念[/color][color=#333333]对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质,而标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示.文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已[1,2],造成错误的原因,可能是有的药品既有对照品,又有标准品.例如,当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品.即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的.[/color]
最近在做呋喃西林ELIA试剂盒,但是没有标准品,我买的sigma的标准品,但是回来需要衍生。不知道以后试剂盒里的标准品是拿我自己衍生好的衍生物做标准品,还是拿sigma的标准物质呢?要么,国内哪里可以买到与我同样的衍生物呢?这个好像比较困难哦,因为不同的人用的衍生方法好像不太一样啊?急盼高人指点哪!!
我做的是生物制品中的氨苄西林残留,内标选的阿莫西林。仪器:美国AB3200,液相是岛津LC-20AD。流动相条件:乙腈,甲酸水(甲酸调PH3.1)梯度程序:0.01min 乙腈5%2min 乙腈5%6min 乙腈80%7min 乙腈5%8min 乙腈5%由于基质里面含有不挥发性盐,所以用了切换阀,前三分钟打进废液,后面再进质谱。这个条件一直做的很好,内标在4min出峰,氨苄西林在5.4min出峰。现在的问题是:条件不能重复了,内标峰形很怪(峰分叉,很毛糙),而且出峰时间延迟了0.5min.而氨苄西林没有变化。如果轻微变一下梯度条件,内标峰就变的很好了,所以我认为质谱是没有问题的。现在的问题就出在液相条件上,我找了很多原因,最开始换了色谱柱,换了两根(同品牌,同规格),一根还是新的,但内标出峰还是一样怪!现在就排除了柱子的问题,那么问题是不是就出在流动相条件上?!乙腈用的牌子是默克的,应该没问题吧。水是制的超纯水,以前也一直这样用的,调PH前是校正了PH仪的,PH值应该还是没问题。但问题还是没有解决!是否是梯度程序问题呢?(但以前一直都是用的这个梯度,重复性很好啊)请教高手,帮忙找下原因啊!这个问题困扰我好久了,方法学已经做完了,现在要测样品了,确出现了这种问题,小妹真的很急啊!
对照品:用于鉴别、检查、含量测定和校正检定仪器性能的标准物质;对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。 标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。 对照品与标准品概念不清?对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已,造成错误的原因,可能是有的药品既有对照品,又有标准品。 例如:当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品。即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的。
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7681612594 七氟醚杂质C Sevoflurane Related Compound C 对照品/标准品1612572 七氟醚杂质 B Sevoflurane Related Compound B 对照品/标准品1612550 七氟醚杂质 A Sevoflurane Related Compound A 对照品/标准品1612540 七氟醚 Sevoflurane 对照品/标准品1612539 盐酸舍曲林 Sertraline Hydrochloride 对照品/标准品1612528 盐酸舍曲林杂质A Sertraline Hydrochloride Related Compound A 对照品/标准品1612517 盐酸舍曲林消旋体混合物 Sertraline Hydrochloride Racemic Mixture 对照品/标准品1612506 L-丝氨酸 L-Serine 对照品/标准品1612426 芝麻油杂质B Sesame Oil Related Compound B 对照品/标准品1612415 芝麻油杂质A Sesame Oil Related Compound A 对照品/标准品1612404 芝麻油 Sesame Oil 对照品/标准品1612029 番泻苷 B Sennoside B 对照品/标准品1612018 番泻苷 A Sennoside A 对照品/标准品1612007 番泻苷 Sennosides 对照品/标准品1611955 硒 蛋氨酸 Selenomethionine 对照品/标准品1611900 盐酸司来吉兰 Selegiline Hydrochloride 对照品/标准品1611004 司可巴比妥 CII Secobarbital CII 对照品/标准品1610090 东莨菪亭 Scopoletin 对照品/标准品1610001 氢溴酸东莨菪碱 Scopolamine Hydrobromide 对照品/标准品1609831 沙奎那韦杂质A Saquinavir Related Compound A 对照品/标准品1609829 甲磺酸沙奎那韦 Saquinavir Mesylate 对照品/标准品1609807 双水杨酯 Salsalate 对照品/标准品1609625 沙美特罗杂质B Salmeterol Related Compound B 对照品/标准品1609614 沙美特罗杂质A Salmeterol Related Compound A 对照品/标准品1609603 昔美酸沙美特罗 Salmeterol Xinafoate 对照品/标准品1609501 水杨酸片 Salicylic Acid Tablets 对照品/标准品1609024 水杨酸杂质B Salicylic Acid Related Compound B 对照品/标准品1609013 水杨酸杂质A Salicylic Acid Related Compound A 对照品/标准品1609002 水杨酸 Salicylic Acid 对照品/标准品1608000 水杨酰胺 Salicylamide 对照品/标准品1607506 连翘粉状贯叶提取物 Powdered St. John's Wort Extract 对照品/标准品1607040 糖精钠 Saccharin Sodium 对照品/标准品1607029 糖精钙 Saccharin Calcium 对照品/标准品1607007 糖精 Saccharin 对照品/标准品1606503 芦丁 Rutin 对照品/标准品1606208 硝酚胂酸 Roxarsone 对照品/标准品1605523 罗哌卡因杂质B Ropivacaine Related Compound B 对照品/标准品1605512 罗哌卡因杂质A Ropivacaine Related Compound A 对照品/标准品1605500 盐酸罗哌卡因 Ropivacaine Hydrochloride 对照品/标准品1604916 罗库溴铵合剂峰的识别 Rocuronium Peak Identification Mixture 对照品/标准品1604905 罗库溴铵 Rocuronium Bromide 对照品/标准品1604870 利凡斯的明杂质B Rivastigmine Related Compound B 对照品/标准品1604869 利凡斯的明杂质A Rivastigmine Related Compound A 对照品/标准品1604814 利托那韦杂质混合物 Ritonavir Related Compounds Mixture 对照品/标准品1604803 利托那韦 Ritonavir 对照品/标准品1604701 盐酸利托君 Ritodrine Hydrochloride 对照品/标准品1604665 利培酮系统适用性试验用混合物 Risperidone System Suitability Mixture 对照品/标准品1604654 利培酮 Risperidone 对照品/标准品1604643 利塞膦酸杂质C Risedronate Related Compound C 对照品/标准品1604632 利塞膦酸杂质B Risedronate Related Compound B 对照品/标准品1604621 利塞膦酸杂质A Risedronate Related Compound A 对照品/标准品1604610 利塞膦酸钠 Risedronate Sodium 对照品/标准品1604600 利美索龙 Rimexolone 对照品/标准品1604508 盐酸金刚乙胺 Rimantadine Hydrochloride 对照品/标准品1604348 利鲁唑杂质A Riluzole Related Compound A 对照品/标准品1604337 利鲁唑 Riluzole 对照品/标准品1604202 醌式利福平 Rifampin Quinone 对照品/标准品1604009 利福平 Rifampin 对照品/标准品1603800 利福布丁 Rifabutin 对照品/标准品1603108 核糖 Ribose 对照品/标准品1603006 维生素B2 Riboflavin (Vitamin B2) 对照品/标准品1602706 利巴韦林 Ribavirin 对照品/标准品1602003 间苯二酚 Resorcinol 对照品/标准品1601849 二类残留溶剂-二甲苯 Residual Solvent Class 2 - Xylenes 对照品/标准品1601827 二类残留溶剂-三氯乙烯 Residual Solvent Class 2 - Trichloroethylene 对照品/标准品1601805 二类残留溶剂-甲苯 Residual Solvent Class 2 - Toluene 对照品/标准品1601780 二类残留溶剂-四氢萘 Residual Solvent Class 2 - Tetralin 对照品/标准品1601770 二类残留溶剂-四氢呋喃 Residual Solvent Class 2 - Tetrahydrofuran 对照品/标准品1601769 二类残留溶剂-二氧噻吩烷 Residual Solvent Class 2 - Sulfolane 对照品/标准品1601747 二类残留溶剂-吡啶 Residual Solvent Class 2 - Pyridine 对照品/标准品1601725 二类残留溶剂-硝基甲烷 Residual Solvent Class 2 - Nitromethane 对照品/标准品1601703 二类残留溶剂-N-甲基吡咯烷酮 Residual Solvent Class 2 - N-Methylpyrrolidone 对照品/标准品1601689 二类残留溶剂-甲基环己烷 Residual Solvent Class 2 - Methylcyclohexane 对照品/标准品1601667 二类残留溶剂-甲基丁基酮 Residual Solvent Class 2 - Methylbutylketone 对照品/标准品1601645 二类残留溶剂- 2-甲氧基乙醇 Residual Solvent Class 2 - 2-Methoxyethanol 对照品/标准品1601623 二类残留溶剂-甲醇 Residual Solvent Class 2 - Methanol 对照品/标准品1601601 二类残留溶剂-己烷 Residual Solvent Class 2 - Hexane 对照品/标准品1601587 二类残留溶剂-甲酰胺 Residual Solvent Class 2 - Formam
本人现在用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测硬脂酸镁,为什么对照品溶液中有很多杂峰,我用的正庚烷是色谱纯级别的,还有到最后计算是用不到对照品的浓度,那为什么还要进对照品?请各位老师指点
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7651724918 唑吡坦杂质A CIV Zolpidem Related Compound A CIV 对照品/标准品1724907 酒石酸唑吡坦 CIV Zolpidem Tartrate CIV 对照品/标准品1724893 唑吡坦 CIV Zolpidem CIV 对照品/标准品1724805 盐酸唑拉西泮 Zolazepam Hydrochloride 对照品/标准品1724769 硫酸锌 Zinc Sulfate 对照品/标准品1724747 氧化锌 Zinc Oxide 对照品/标准品1724689 齐留通杂质C Zileuton Related Compound C 对照品/标准品1724678 齐留通杂质B Zileuton Related Compound B 对照品/标准品1724667 齐留通杂质 A Zileuton Related Compound A 对照品/标准品1724656 齐留通 Zileuton 对照品/标准品1724532 齐多夫定杂质C(胸腺嘧啶) Zidovudine Related Compound C (thymine) 对照品/标准品1724521 齐多夫定杂质B Zidovudine Related Compound B 对照品/标准品1724500 齐多夫定 Zidovudine 对照品/标准品1724317 扎西他滨杂质A Zalcitabine Related Compound A 对照品/标准品1724306 扎西他滨 Zalcitabine 对照品/标准品1724000 盐酸育亨宾 Yohimbine Hydrochloride 对照品/标准品1722005 木糖 Xylose 对照品/标准品1721002 盐酸赛洛唑啉 Xylometazoline Hydrochloride 对照品/标准品1720600 木糖醇 Xylitol 对照品/标准品1720429 盐酸赛拉嗪 Xylazine Hydrochloride 对照品/标准品1720407 赛拉嗪 Xylazine 对照品/标准品1720203 呫吨酮 Xanthone 对照品/标准品1720000 呫吨酸 Xanthanoic Acid 对照品/标准品1719102 华法林杂质 A Warfarin Related Compound A 对照品/标准品1719000 华法林 Warfarin 对照品/标准品1717708 牡荆素(牡荆甙) Vitexin 对照品/标准品1717504 含量测定系统适用性用维生素D Vitamin D Assay System Suitability 对照品/标准品1716002 维生素A Vitamin A 对照品/标准品1715000 硫酸紫霉素 Viomycin Sulfate 对照品/标准品1714528 长春瑞滨杂质A Vinorelbine Related Compound A 对照品/标准品1714506 酒石酸长春瑞滨 Vinorelbine Tartrate 对照品/标准品1714007 硫酸长春新碱 Vincristine Sulfate 对照品/标准品1713004 硫酸长春碱 Vinblastine Sulfate 对照品/标准品1711508 阿糖腺苷 Vidarabine 对照品/标准品1711472 维替泊芬杂质A Verteporfin Related Compound A 对照品/标准品1711461 维替泊芬 Verteporfin 对照品/标准品1711440 维拉帕米杂质F Verapamil Related Compound F 对照品/标准品1711439 维拉帕米杂质E Verapamil Related Compound E 对照品/标准品1711428 维拉帕米杂质D Verapamil Related Compound D 对照品/标准品1711406 维拉帕米杂质B Verapamil Related Compound B 对照品/标准品1711304 维拉帕米杂质A Verapamil Related Compound A 对照品/标准品1711224 维库溴铵杂质F Vecuronium Bromide Related Compound F 对照品/标准品1711202 盐酸维拉帕米 Verapamil Hydrochloride 对照品/标准品1711188 维库溴铵杂质C Vecuronium Bromide Related Compound C 对照品/标准品1711177 维库溴铵杂质B Vecuronium Bromide Related Compound B 对照品/标准品1711166 维库溴铵杂质A Vecuronium Bromide Related Compound A 对照品/标准品1711155 维库溴铵 Vecuronium Bromide 对照品/标准品1711133 赖氨加压素 Lypressin 对照品/标准品1711100 加压素 Vasopressin 对照品/标准品1711009 香草醛熔点标准品 Vanillin Melting Point Standard 对照品/标准品1710006 香草醛 Vanillin 对照品/标准品1709018 Vancomycin B with Monodechlorovancomycin 对照品/标准品1709007 盐酸万古霉素 Vancomycin Hydrochloride 对照品/标准品1708795 缬沙坦杂质 C Valsartan Related Compound C 对照品/标准品1708784 缬沙坦杂质 B Valsartan Related Compound B 对照品/标准品1708773 缬沙坦杂质 A Valsartan Related Compound A 对照品/标准品1708762 缬沙坦 Valsartan 对照品/标准品1708751 戊柔比星分离度用混合物 Valrubicin Resolution Mixture 对照品/标准品1708730 戊柔比星 Valrubicin 对照品/标准品1708729 丙戊酸杂质A Valproic Acid Related Compound A 对照品/标准品1708718 丙戊酸杂质 B Valproic Acid Related Compound B 对照品/标准品1708707 丙戊酸 Valproic Acid 对照品/标准品1708503 L- 缬氨酸 L-Valine 对照品/标准品1708015 D-缬更昔洛韦 D-Valganciclovir 对照品/标准品1708004 缬更昔洛韦盐酸盐 Valganciclovir Hydrochloride 对照品/标准品1707908 缬草烯酸 Valerenic Acid 对照品/标准品1707894 万乃洛韦杂质G Valacyclovir Related Compound G 对照品/标准品1707883 万乃洛韦杂质F Valacyclovir Related Compound F 对照品/标准品1707872 万乃洛韦杂质E Valacyclovir Related Compound E 对照品/标准品1707861 万乃洛韦杂质D Valacyclovir Related Compound D 对照品/标准品1707855 万乃洛韦杂质C Valacyclovir Related Compound C 对照品/标准品1707839 盐酸万乃洛韦 Valacyclovir Hydrochloride 对照品/标准品1707806 熊去氧胆酸 Ursodiol 对照品/标准品1706701 C13尿素 Urea C 13 对照品/标准品1706698 尿素 Urea 对照品/标准品1706009 乌拉莫司汀 Uracil Mustard 对照品/标准品1705800 阿糖尿苷 Uracil Arabinoside 对照品/标准品1705505 十一烯酸 Undecylenic Acid 对照品/标准品1705323 泛癸利酮杂质A Ubidecarenone Related Compound A 对照品/标准品1705312 系统适用性试验用泛癸利酮 Ubidecarenone for System Suitability 对照品/标准品1705301 泛癸利酮 Ubidecarenone 对照品/标准品1705006 L- 酪氨酸 L-Tyrosine 对照品/标准品1704003 泰洛沙泊 Tyloxapol 对照品/标准品1703850 酒石酸泰洛星 Tylosin Tartrate 对照品/标准品1703805 泰洛星 Tylosin 对照品/标准品1702008 氯筒箭毒碱 Tuboc
[b]Q:[b][b][b][/b][/b]阿莫西林克拉维酸钾颗粒的检测,应用编号是?(因发题失误,今天答题时间延长至4点30分)[/b]A:103591(因为出题失误,今天回答一律正确)===============================================================【活动内容】1、每个工作日上午10:00左右发布一个关于应用数据库的应用问答题,版友根据题目给出自己理解的答案。2、每个工作日下午15:10公布参考答案。【活动奖励】幸运奖:抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励[color=#ff0000]2钻石币[/color](抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);中奖名单:吕梁山(注册ID:shih20j07)WUYUWUQIU(注册ID:wulin321)zimeng3211(注册ID:zimeng3211)zengzhengce163(注册ID:zengzhengce163)lijing320323(注册ID:lijing320323)[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131632000260_5567_1610895_3.png!w690x388.jpg[/img][img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131631572757_528_1610895_3.png!w690x388.jpg[/img]积分奖励:所有回答正确的版友奖励[color=#ff0000]10个积分[/color](幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次[/b][align=left][color=#ff0000][b]PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。[/b][/color][/align][align=left][color=#ff0000][b] 下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。[/b][/color][/align][align=center]=======================================================================[/align]方法:HPLC基质:药品应用编号:103591化合物:阿莫西林样品前处理:1、对照品溶液:阿莫西林 (0.8 mg/mL) 流动相溶解。2、供试品溶液:取样品适量,加水溶解,使阿莫西林含量为0.5 mg/mL,过滤,取滤液。色谱条件:色谱柱: Diamonsil C18(2) 150*4.6 mm,5 μm(Cat#:99901)流动相: 0.05mol/mL磷酸二氢钠溶液(7.8 g磷酸二氢钠加900mL水,10%氢氧化钠或磷酸调pH=4.4)-甲醇=95:5流速: 1.0 mL/min柱温: 30 ℃检测器: UV 220nm进样量: 20 μL文章出处:天津应用实验室关键字:阿莫西林克拉维酸钾颗粒、阿莫西林、2010药典、Diamonsil C18(2)、HPLC摘要:Diamonsil C18(2)检测阿莫西林克拉维酸钾颗粒中阿莫西林。图谱:[img]http://www.dikma.com.cn/u/image/2015/08/03/1438588907674178.png[/img][img]http://www.dikma.com.cn/u/image/2015/08/03/1438588916104003.png[/img]

10,抽取5个版友);中奖名单:千层峰(注册ID:jxyan)大川之子,纵横四海(注册ID:chuangu120)m3071659(注册ID:m3071659)莫名其妙(注册ID:moyueqiu)sunpengwjh(注册ID:sunpengwjh)http://ng1.17img.cn/bbsfiles/images/2016/09/201609281518_612452_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609281518_612453_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================美洛昔康片方法:HPLC基质:药品应用编号:101386化合物:美洛昔康固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil C18(2) 5u 150 x 4.6mm样品前处理:【有关物质】 取本品细粉,加碱性甲醇溶液(取40%甲醇溶液100 ml,加0.4 mol/L氢氧化钠溶液6 ml,混匀)溶解并稀释成每1 ml中约含有1 mg的溶液,滤过,取续滤液作为供试品溶液;精密量取1 ml,置100 ml量瓶中,用上述碱性甲醇溶液稀释至刻度,摇匀,作为对照溶液。色谱条件:检测器:UV 270 nm 流动相:0.1 mol/L 乙酸铵溶液-甲醇(50:50) 进样量:20 ul文章出处:P611关键字:美洛昔康,2010版中国药典,HPLC,含量测定,钻石二代,Diamonsil C18(2)谱图:http://www.dikma.com.cn/Public/Uploads/images/meiluoxikang.GIF
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]新手,请教各位老师一个问题,最近做呋喃西林,waters tqd [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]ms,调谐是峰比较大,就是正常,浓度0.5,调谐后进样总是没峰出,首先流动相洗脱能力和时间肯定够,对照浓度用了调谐的浓度了,标准是纳克,以前做是有峰的,现在不知道什么原因?求教

公司需要进行环氧乙烷的残留检测,我负责进行外标法标准曲线的制作以及仪器参数的探索。由于公司未配置自动进样器,所以在此项目中我们是使用顶空手动进样。这对于初次进行该试验的人员来说无疑加大了各项难度,其影响便体现在试验结果的平行性与重复性上。前期经过几次的试验,对同一样品的测试结果都远达不到要求,更不用谈标准曲线的制作了。后来在论坛里看到爱吉仁产品的试用活动,便申请了些西林瓶试用。一周多之后到货,打开包装,有10mL,20mL的螺口和钳口的西林瓶及对应瓶盖。以及其他样品瓶,不过在本次的试验中暂未使用到,主要谈一下西林瓶的使用体会。http://ng1.17img.cn/bbsfiles/images/2017/10/2015082109080701_01_0_3.jpg 在试验中我选用的是螺口瓶,因为比较方便,而且密封效果也还不错。但是就瓶子的本身质量来说,似乎比之前我使用过的稍微薄了些。大致说一下前处理实验的流程:手动顶空进样,1mL气密性注射针,样品在恒温水浴锅内进行气液平衡后开始实验。色谱柱Agilent DB-624,仪器岛津GC-2014。在之前的实验中,同一样品多次进样检测,所得样品峰越来越小,直至几乎无峰出现。可能原因主要有两方面:一是仪器本身的问题,二是样品的前处理过程出现问题,包括进样的操作以及准确性。后对仪器进行了验证,使用纯乙醇进样走谱,可得到完好峰形,见下图。排除原因一。http://ng1.17img.cn/bbsfiles/images/2017/10/2015082109232557_01_2699629_3.jpg那么很大可能是样品的处理过程出现了操作差错。之后重新对样品处理,使用了爱吉仁10mL螺口西林瓶,对环氧乙烷标准液进行加热至气液平衡,检测。共检测5组10个样品,用于绘制标准曲线。参见下图单一样品谱图及环氧乙烷标准曲线图和相关信息。因无法上传,谱图详见附件。由数据分析可知,标准曲线的制作仍难达标,在舍弃部分误差较大数据后所得的曲线勉强可用,后期将继续优化改进。http://ng1.17img.cn/bbsfiles/images/2015/08/201508211406_561911_2989334_3.jpgEO(外标法)标准曲线图http://ng1.17img.cn/bbsfiles/images/2015/08/201508211406_561912_2989334_3.jpg环氧乙烷检测谱图ahttp://ng1.17img.cn/bbsfiles/images/2015/08/201508211406_561913_2989334_3.jpg环氧乙烷检测谱图bhttp://ng1.17img.cn/bbsfiles/images/2015/08/201508211406_561914_2989334_3.jpg样品前处理







 我要推广仪器
我要推广仪器
 下载APP
下载APP