清华胡泽平团队揭示代谢组学结合AI模型在胃癌诊断及预测患者预后中的临床应用潜能
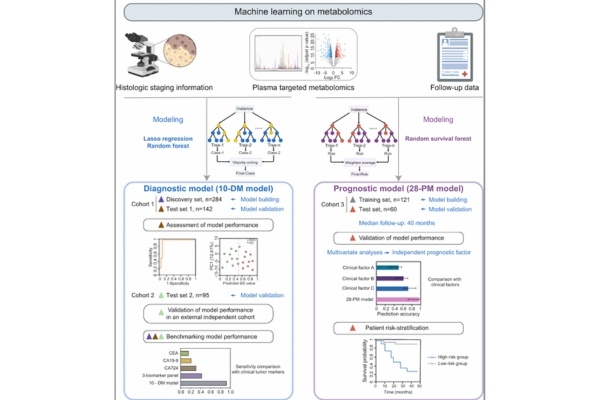
2024年2月23日,清华大学药学院胡泽平团队与合作者在《Nature Communications》发表题为“Metabolomic machine learning predictor for diagnosis and prognosis of gastric cancer”的研究论文,揭示了胃癌患者血浆的代谢重编程图谱,并发现基于代谢组学构建的机器学习模型能准确诊断胃癌患者,并预测患者预后风险。 研究背景 胃癌是东亚地区高发的致死性肿瘤。胃癌的早期确诊和及时干预对改善临床结果至关重要。然而,作为胃癌诊断金标准的内镜活检因其侵入性高且费用昂贵而限制了临床应用。因此,迫切需要开发具有高灵敏度和特异性的新型非侵入性胃癌检测方法。此外,对病人进行预后监测并及时进行干预有助于更好的临床结果。因此,开发一种更精确的患者预后预测方法至关重要。胃作为具有高度异质性的消化器官,其癌变和进展受到基因突变和环境扰动的双重影响,伴随显著的代谢重塑。然而,代谢重编程及其在胃癌诊疗中的潜在应用并未被系统性深入地研究过,未能满足临床对胃癌早诊和患者分层治疗的需求。目前的胃癌生物标志物研究很大程度上受限于队列规模小、缺乏独立的验证队列、样本类型和检测方法的差异导致的结果再现性低,以及受分析技术限制的检测灵敏度有待提高等问题。因此,使用多中心、大队列、特征明确的胃癌和对照人群进行代谢组学分析对于识别和验证具有转化潜力的生物标志物,从而开发和完善适合临床应用的代谢生物标志物的仍然势在必行。 研究过程 研究人员收集了702例胃癌患者和非胃癌对照的血浆样本,进行了靶向代谢组学数据分析。结果显示,胃癌患者血浆发生了明显的代谢重编程,其中最显著改变的代谢通路为谷胱甘肽代谢。通路中的两种关键代谢物 还原型谷胱甘肽GSH 和氧化型谷胱甘肽 GSSG 在胃癌血浆中显着降低。此外,作为氧化应激紊乱指示物的GSH/GSSG 比率在胃癌患者中显着上调,并随着疾病进展而逐渐增加。表明胃癌患者血浆中氧化应激严重失调。此外,胃癌患者的半胱氨酸和蛋氨酸代谢通路也发生显著失调。与非胃癌对照相比,胃癌患者的 S-腺苷-L-同型半胱氨酸 (SAH) 下调,S-腺苷甲硫氨酸 (SAM) 上调,并且 SAM/SAH 比值随疾病进展而增加。作为通用甲基供体,SAM 丰度和SAM/SAH 比值的失调可能反映了胃癌患者甲基池的扰动。这些胃癌血浆中的代谢重编程特征为开发胃癌检测和患者预后预测生物标志物奠定了基础。图1. 本研究设计及流程图尽管代谢组学在全面分析胃癌整体代谢特征方面具有独特的优势,能够大规模识别用于 GC 诊断和预后的有希望的生物标志物,但复杂的组学数据的解释始终是一个挑战。在过去的几年中,机器学习算法已被用于发现组学数据和疾病状态之间的潜在关联并创建预测模型。因此,研究人员分别使用随机森林和随机生存森林算法建立了基于10个代谢物的胃癌诊断模型(10-DM)和基于28个代谢物的胃癌患者预后预测模型(28-PM)并在测试集中验证了模型的优越性能。对模型效果评估时发现,10-DM诊断模型即使对早期胃癌患者(stage IA)也能准确诊断,表现出比临床正在使用的癌症蛋白标志物CEA,CA19-9,CA72-4等更优越的诊断效果(灵敏度0.925:0.428)。10-DM模型的准确性和重现性在覆盖521人的多中心队列中得到证实,表明该模型具有较高的稳健性和临床应用潜力。此外,28-PM预后模型比利用临床参数的传统模型的预测效果更好(C-index值0.816:0.591),并能有效地将患者分为高低两个风险组。在中位数为40个月的随访期间,28-PM 模型区分的高风险患者的预后与低风险患者相比更差,证明了模型的预测能力。被分层为高危险组的患者更有可能受益于强化监测、及时干预和新型治疗药物的试验。 研究结果 综上,该研究描述了胃癌患者血浆的整体代谢重编程,并结合机器学习算法构建了两个模型,分别识别胃癌患者并预测其预后。该工作有助于进一步理解胃癌的分子病理学特征,促进了胃癌早期检测的发展,并为实现胃癌的精准治疗提供理论基础。迄今为止报道的胃癌组学研究主要集中在探究以 DNA、RNA 和蛋白质作为胃癌生物标志物的潜力,而该工作强调了胃癌中循环代谢物的预测价值。通过使用高灵敏代谢组学技术分析覆盖共计702例胃癌和非胃癌对照的多中心样本已经独立测试集的设定,该研究成功应对了生物标志物探究工作普遍面临的结果再现性低,无法进行临床推广应用的挑战。未来可以通过建立靶向两个模型中代谢物的特定子集的靶向定量代谢组学检测方法以提高效率并降低成本,并在来自更多中心的更大规模临床样本中进行验证和优化。此外,基于这两种预测模型有望促进胃癌无创早期检测,并根据患者的风险分层为临床决策提供信息,从而实现辅助胃癌精准诊疗策略的临床转化。胡泽平 清华大学个人简介:分别于山东大学齐鲁医学院、中国食品药品检定研究院和新加坡国立大学获医学学士、药理学硕士和Ph.D.学位。后于美国西北太平洋国家实验室Richard D. Smith组从事生物质谱和代谢组学的博士后研究。2012年受聘于美国德克萨斯大学西南医学中心任研究助理教授、儿童研究所代谢组学平台技术主任。2016年12月起任清华大学药学院准聘系列PI、特别研究员,2024年1月任长聘副教授。研究方向为“基于新型代谢组学/多组学技术研发的疾病代谢重塑研究、新药靶标与生物标志物发现”,包括:1)肿瘤微环境中不同类型细胞(特别是神经细胞/神经递质与肿瘤细胞和免疫细胞间)的代谢互作与单细胞代谢异质性、功能与代谢调控分子机制解析,与新药靶标发现;2)心血管疾病的代谢重塑规律、功能、调控分子机制解析,与新药靶标发现;3)超灵敏、单细胞代谢组学技术,及基于AI的多组学数据智能化整合分析技术与大模型研发;近年来以通讯作者(含共同)在Cell Metabolism (2018), Nature Metabolism (2021a 2021b), Nature Cancer (2022), Science Translational Medicine (2018), Journal of Clinical Investigation (2022), Nature Cardiovascular Research (2022), Nature Communications (2024 2021a 2021b), Cancer Research (2024), Cell Discovery (2022), Analytical Chemistry (2021)等期刊发表论文多篇。获邀在Nature Metabolism (2023), TrAC Trends in Analytical Chemistry (2023), Acta Pharmaceutica Sinica B (2023), Pharmacology & Therapeutics (2021), Clinical Pharmacology & Therapeutics (2019)等期刊发表Viewpoints或综述,共已发表论文60余篇,引用8000余次(Google scholar),H-index为41。研究成果多次被Science, Nature Cancer, Nature Reviews Cancer等期刊作为研究亮点专评。先后主持/参与国家基金委面上项目、重大研究计划重点项目、集成项目、“未来生物技术”原创探索项目;科技部国家科技重大专项、重点研发专项(2项)等共7项国家级科研项目;及国际头部药企资助的新药研发合作项目。担任国家基金委项目会评专家,Nature Metabolism, Nature Communications, Science Advances, Cell Reports等多个期刊审稿人,及Life Metabolism, Acta Pharmaceutica Sinica B等期刊编委。























 我要推广仪器
我要推广仪器
 下载APP
下载APP