第一步:甲氧基聚乙二醇的合成聚乙二醇在无水二氯甲烷中与金属钠作用生成聚乙二醇钠, 然后与碘甲烷反应即得。一甲氧基聚乙二醉、双端都反应的二一甲氧基聚乙止醇和未反应的聚乙二醇的反应混合物硅胶柱层析色潜提纯可以得到纯净的甲氧基聚乙二醇第二步:甲氧基聚乙二醇丁二酸单醋的合成将甲氧基聚乙二醇(Me-PEG-2000)、丁二酸酐和催化剂加入盛有二氯甲烷的圆底烧瓶中, 磁力搅拌使固体完全溶解后, 室温搅拌反应过夜。反应液分别用盐酸水溶液、氢氧化钠水溶液和甲醇水溶液依次洗涤。有机相经无水MgSO4干燥, 过滤除去干燥剂, 减压蒸除有机溶剂, 残留物以石油醚结晶, 收率90%。第三步:甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺的合成甲氧基聚乙二醇丁二酸单酷先经N一羟基丁二酰亚胺(NHS)活化, 然后缓慢滴加人到二硬脂酰磷脂酰乙醇胺(DSPE)的三氯甲烷中, 加料完毕后继续反应4h, 蒸除溶剂, 浓缩液在乙醚中结晶,硅胶柱层析色谱提纯可以得到自色粉末状固体的。甲氧基聚乙二醇一二硬脂酰磷脂酰乙醇胺。来源:中国标准物质网
请问1,2-丙二醇单甲醚和甲氧基丙酮混合物中甲氧基丙酮的含量如何分析?谢谢!
本人在用NY/T 1380-2007用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]作51种农药的分析时,发现里面要用所谓的保护剂即:3-乙氧基1,2-丙二醇,想在这里请教一下,这东西到底怎么个保护法?不加可以吗?会有什么样的影响 ?请各位作过的老师们指导一下。先谢谢了。
求助2-甲氧基乙醇 2-乙氧基乙醇的详细测试方法,用什么溶剂,特征离子峰,谢谢!
我们是做化妆品的企业,因为产品中有添加辛二醇、辛氧基甘油作为防腐剂,现需要检测其含量。但我们用甲醇加样品溶解后用HPLC测试却没有跑出任何峰出来,想请问一下各位高手:1.这二类物质是否可以用HPLC来检测? 2.是否我们的样品处理方法有问题? 3.对于不出峰的样品,大家是否有其他的一些样品处理方法或测试条件等各方面的建议?? 急!急!!急!!!急!!!!急!!!!![em0705]
q请问分析氯乙氧基乙醇用什么柱好?是进口的,国产的,还是自己装的+
有做聚乙氧基化非离子表面活性剂中聚乙二醇含量的测定 高效液相色谱法 的没有,请教下外标法做曲线,聚乙二醇标样600-2000,是只取一种配制不同浓度,做曲线,还是600,1000,2000的都取。具体怎么个情况?
各位大虾好,小第有一疑问盼大家给点意见。我做美国药典中的盐酸米托蒽醌中的残留溶剂时,按照厂家给的资料要检测其中的2-甲氧基乙醇,厂家给的方法为直接进样(水做溶剂)。但它的样品浓度为200mg/ml,按照盐酸米托蒽醌的溶解度看,样品是绝对不能完全溶解的,样品溶解后成米糊状,离心处理也处理不到上清液,进样发现样品出峰非常的难看,且不见残留溶剂峰。我们打算自己开发方法用顶空做,但美国药典467中建议2-甲氧基乙醇用直接进样法,各位大虾你有过内似的经历么,2-甲氧基乙醇用顶空法检测后,美国FDA能批准么?
主峰是3-薄荷氧基-1,2-丙二醇(即MOPD),现在想了解其他几个小杂峰是什么结构,请大家帮忙看看。主要是11.116,12.93,13.556,14.025这四个,谢谢大家
请问十二烷氧基葡糖苷、十二烷基葡糖苷、聚乙二醇葡糖苷性能有什么不同?
请问一下正己基三乙氧基硅烷,自动进样针的清洗溶剂用什么合适,我用乙醇老是堵????太费针了
大家新年好!请问,水中如果可能含有2-丁氧基乙醇,想定性,能不能直接进[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱联用仪呢?如果不能应该用什么溶剂提取出来测定呢?多谢多谢哦!
大家谁有对甲氧基苯乙醇,麻烦打个质谱图让我看看,我现在在做一个反应,产物质谱图不确定,谢谢!
请教:乙烯基甲醚、丙烯醛、二甲氧基二氢吡喃、戊二醛这几种物质可否有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?用什么样的柱子和检测条件?
标准号: GB/T 17830-1999 中文标题: 聚乙氧基化非离子表面活性剂中聚乙二醇含量的测定 高效液相色谱法 英文标题: Determination of polyethylene glycol in polyethoxylated nonionic surfactants by HPLC 文摘: 本标准规定了测定聚乙氧基化非离子表面活性剂中聚乙二醇(PEG)含量的高效液相色谱(HPLC)法。本标准适用于测定聚乙氧基化脂肪醇和聚乙氧基化烷基酚中聚乙二醇的含量。 发布日期: 1999-8-12 实施日期: 2000-2-1 废止日期: 被替代标准: 引用标准: 采用关系: 开本页数: 6 中标分类号: G72 ICS号: ICS 71.100.40 发布单位: 国家质量技术监督局 或者有醇的水溶液中的聚乙二醇20000,10000,6000的测定方法。应助者,我给积分。
徐建强(南京气象学院环境科学系,南京 210044)分光光度法是获得物质光吸收特性及定性、定量分析的重要手段,在冶金、地质、生物、医学、农业、环境监测、食品卫生等部门得到极其广泛的应用。分光光度法的发展不仅依赖于电子学、激光和计算机技术的发展和应用,而且还依赖于高灵敏度、高选择性有机试剂的合成和应用。喹啉类试剂作为光度分析的有机试剂,可以分为两类,一类是喹啉及其衍生物,如8-羟喹啉、8-巯基喹啉、8-氨基喹啉等,这类试剂可用作金属离子光度分析的显色剂,具有一定的灵敏度和选择性 另一类是喹啉偶氮化合物,喹啉偶氮化合物作为一大类显色剂已有多种试剂被合成和研究。其中8-氨基喹啉的8位偶氮衍生物前苏联学者研究得较早 我国的李亚文等也进行了系统深入的研究,近年来不断有一系列新的衍生物合成,这些化合物因其特有的灵敏度和选择性而备受化学工作者的关注。自从1955年程广禄等[3]首次提出PAN(即1-(2-吡啶偶氮)-2-萘酚)作为分析试剂后,越来越多的偶氮试剂相继被化学工作者合成并且用于光度分析研究。实践证明,偶氮化合物(azo-compounds)具有性质稳定、显色反应灵敏度高、选择性好、对比度大等优点,仍然是目前应用最广泛的一类显色剂。本文选用6-甲氧基-8-氨基喹啉作为原料,首先对其进行重氮化,得到重氮盐,然后与间苯二酚进行偶联反应,合成了新有机试\剂:4-(6-甲氧基-8-喹啉偶氮)-间苯二酚(简称MQAR)。对其进行了结构分析和鉴定。实验研究表明,MQAR能与Co、Cu、Fe、Ni等金属离子发生灵敏的显色反应,该试剂可用于试样中微量金属离子的测定(另文介绍),方法简便、快速、准确可靠,是一种比较理想的新有机试剂。1 合成方法1.1 试剂和仪器设备6-甲氧基-8-氨基喹啉(由工业品提纯所得),亚硝酸钠(AR),间苯二酚(AR),N,N-二甲基甲酰胺(AR),浓硫酸(AR),甲酸(AR)。中量有机化学制备仪、真空干燥箱、差热分析仪、Perkin-Elmer元素分析仪、IR-408型红外分光光度计、756MC型紫外-可见分光光度计、BRUKERARX300M核磁共振谱仪。1.2 合 成1.2.1 合成线路MQAR的合成线路如图1所示。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221937_18814_1634962_3.gif[/img]1.2.2 合成步骤(1)重氮化 在250mL三口烧瓶中,加入8.7g6-甲氧基-8-氨基喹啉,10mL甲酸,同时加入由15mL浓硫酸和10mL水配制而成的溶液,搅拌溶解,置于冰浴中冷却。在0~5℃时边搅拌边滴加3.5g亚硝酸钠与10mL水配制而成的溶液,控制在1h内加完,使反应充分完全,最后得到深红色的重氮盐溶液,用于下一步的反应。(2)偶联 取5.5g间苯二酚溶于75mL无水乙醇中,置于冰浴中冷却。在0~5℃时,边搅拌边加入上述重氮盐溶液,控制重氮盐溶液于1h内加完,继续搅拌2h,静置过夜得砖红色沉淀,抽滤,依次用水、无水乙醇洗涤,抽干。干燥后得棕黄色粗品。(3)精制 将偶联得到的粗品用N,N-二甲基甲酰胺重结晶两次,于真空干燥箱干燥,得到深红色的MQAR纯品。经测定产品熔点为194℃。2 结构分析2.1 薄层色谱分析(1)试剂和仪器设备 展开剂正丁醇∶无水乙醇∶2molL-1氨水=3∶1∶1 支持剂硅胶HF254+0.3%CMC 8×10cm2薄层板(自制) 层析缸。(2)结果和讨论 在不同极性的溶剂体系中进行展开,在薄层色谱板上只发现一个斑点,在紫外灯光下未发现其他斑点,(1)中所列展开剂效果最好,比移值Rf=0.66(图2)。结果表明,产品中只含有一种物质。2.2 元素分析用Perkin-Elmer元素分析仪对产品进行了元素分析,产品MQAR元素分析结果与理论计算值基本一致。2.3 紫外-可见吸收光谱分析用756MC型紫外-可见分光光度计测得2×10-5molL-1MQAR的10%DMF水溶液(pH=8.3时)的紫外-可见吸收光谱(图3)。Kmax=450nm,E=2.98×104Lmol-1cm-1。结果表明,产品分子是一个大的共轭体系。2.4 红外吸收光谱分析用IR-408型红外分光光度计对产品MQAR进行了红外吸收光谱分析(KBr压片法),产品的红外光谱解析结果见表1。解析结果表明,产品分子中含有酚羟基、芳环、偶氮基等官能团,还具有1,2,4-三取代苯结构。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221954_18815_1634962_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18816_1634962_3.gif[/img]2.5 核磁共振谱分析用BRUKERARX300M核磁共振谱仪(DMSO-d6溶剂,TMS内标)对产品进行核磁共振谱分析,得到化学位移、峰面积等(表2)。解析结果表明,产品分子中除了羟基质子以外,还含有甲氧基质子以及苯环和杂环质子。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18817_1634962_3.gif[/img]2.6 产品的结构根据实验条件以及结构鉴定和分析,可以确定产品4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的分子结构为[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221956_18818_1634962_3.gif[/img]3 结 论研究了新有机试剂4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的合成方法、实验条件和精制方法,通过薄层色谱法、元素分析、紫外-可见吸收光谱法、红外吸收光谱法和核磁共振谱法等分析手段,对合成的产品进行了分析和结构鉴定,实验结果确证合成了4-(6-甲氧基-8-喹啉偶氮)-间苯二酚纯品。
谁知道微量乙氧基喹啉怎么测定,我好着急呀,哪为仁兄可以告诉我吗
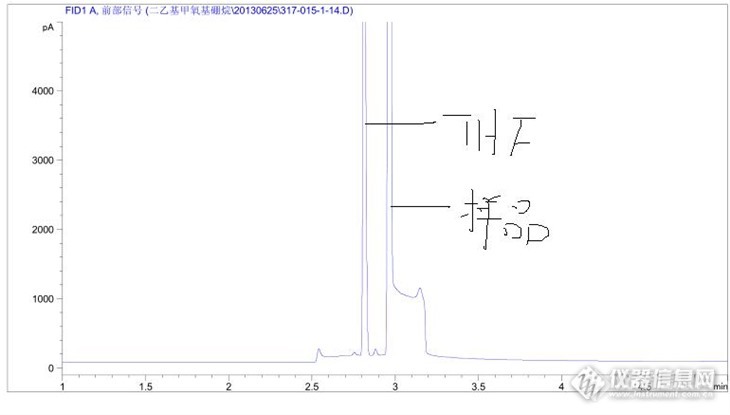
二乙基甲氧基硼烷的THF(四氢呋喃)溶液,有少量甲醇和乙醚。FID检测器HP-5的色谱柱 80℃1分钟 10℃/min 升至180℃ 2分钟以前做的挺好,但是最近成这样了,是什么原因啊?换了新柱子还是这样。http://ng1.17img.cn/bbsfiles/images/2013/07/201307051012_449530_2260586_3.jpg这是以前做的http://ng1.17img.cn/bbsfiles/images/2013/07/201307051012_449531_2260586_3.jpg这是现在的
[size=4][color=#DC143C][font=黑体]同位素比质谱方法检测内源性类固醇雄烯二酮[/font][/color][/size]=========================================在所使用的禁用物质中,类固醇激素是较为普遍使用的一类药物。人体自身能合成与分泌的类固醇激素称为内源性类固醇激素,如攀酮。由于在检测方法上有一定难度,一些运动员选择使用内源性类固醇制剂以逃避兴奋剂检测。目前,兴奋剂检测实验室应用同位素比质谱分析方法检测内源性类固醇来源。13C和12C是碳元素在自然界中的天然同位素。有机化合物的来源不同,其同位素比(如13C与12C的比值)也不同。人体自身分泌的类固醇与相同化学结构的类固醇制剂的同位素比不同。应用同位素比质谱分析技术可以测定化合物13C与12C的比值,同位素比用δ(‰)值表示。根据仪器的分析流程和组成部分,本文方法称为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比质谱([url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS)方法。该方法在兴奋剂检测中的应用时间较短,文献方法较为繁琐,本文建立了快速灵敏的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析方法。-------------------------------------------------试验材料与方法1. 试剂和对照品试剂:β-葡萄糖醛酸酶,Sigma;其余均为国产分析纯试剂。对照品:睾酮(缩写T)、雄酮(缩写An)、本胆烷醇酮(缩写Etio)、5α-雄烷-3α,17β-二醇(缩写5α-diol)、5β-雄烷-3α,17β-二醇(缩写5β-diol)、孕二醇(缩写PD)购自Sigma公司。2. 样品两名健康志愿者,一名男性40岁,尿样为sample 1;一名女性38岁,尿样为sample 2,均没有服用任何药物。收集其晨尿为阴性对照尿。阳性尿样为世界反兴奋剂机构水平考试所用尿样来自兴奋剂检测中心。3. 仪器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比值质谱仪(HP6890/DELTA PLUS,Finnigan);高效液相色谱仪(Waters2796,检测器:Waters2996 PAD,自动收集器:Waters Fraction Collector Ⅲ);Anilent 5973i[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用仪。4. 方法4.I [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS操作条件色谱柱:HP5毛细管色谱柱,25m×0.2mm i.d.×0.3μm;柱流速:1mL/min(室温);升温程序:60 ℃(2min)一50 ℃/min→255℃一2.5℃/min→280℃(6.5min);进样口温度:260℃;燃烧炉温度:960℃;质谱离子源:EI;参考气:CO2,1.8V。4.2 高效液相色谱仪操作条件色谱柱:ZQRBAX SB-C18(4.6mm×250mm,5μm);流动相:水-乙睛,梯度洗脱(0-18min:乙睛从30%→100%);流速1mL/min;柱温:室温。4.3 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD操作条件色谱柱:HP-1毛细管色谱柱,25 m×0.2mm i.d.×0.11μm;柱流速:1 mL/min (室温);升温程序:150 ℃(1min一5℃/min→200℃一30℃/min→310℃(10min)。4.4 样品预处理取尿样2mL,加人1Ml0.2mol pH=7.0的磷酸盐缓冲液和10μLβ-葡萄糖醛酸酶(5000 IU)混匀,在55℃恒温水浴中培养3h,取出后加pH=8.8的碳酸盐固体缓冲剂约100mg 和5mL叔丁基甲醚,振荡萃取,离心后,取出上层有机溶液,在加热的情况下,用氮气吹干,加人50μL甲醇溶解残渣,备用。将上述甲醇溶液置HPLC仪上,依前述色谱条件分离,分段收集流出液,确定收集时间程序。分别将流出组分用氮气吹干,加人50μL环己烷,备用。4.5 对照品溶液的制备分别配制对照品睾酮、雄酮、本胆烷醇酮、5α-雄烷-3α,17β-二醇、5β-雄烷-3α,17β-二醇,孕二醇的甲醇溶液,浓度为1mg/mL。4.6 样品测定4.6.1 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD 分析依上述[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD仪器操作条件,取样品溶液及对照品溶液进行全扫描,扫描范围m/z 20~450,选择待测物的特征离子,获得SIM图。经对样品中与对照品有相同保留时间的峰进行质谱分析,及与标准品质谱图的对比,确定待测样品的组成。4.6.2 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析依上述CC/C/IRMS仪器操作条件,对处理后的样品进行[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析,测得内源性类固醇激素的δ值。
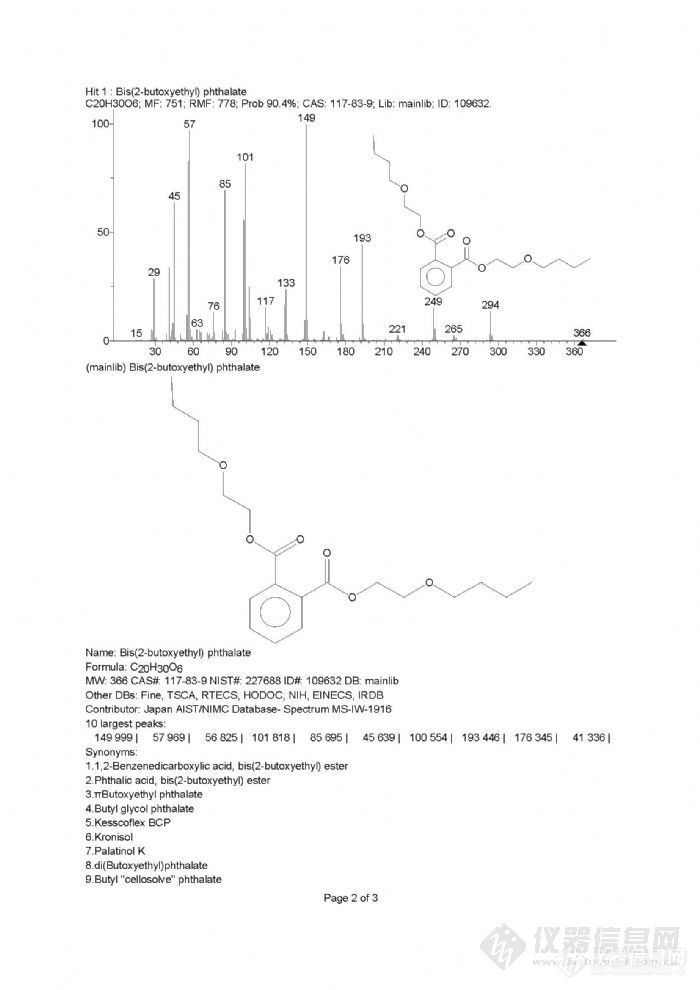
GB/T 21911-2008 《食品中邻苯二甲酸二甲酯的测定》中说明邻苯二甲酸二丁氧基乙基酯(DBEP)的特征离子为149(100) 223(14) 205(9) 278(3),但我进的标准品丰度与标准差很多。下图为我的DBEP标准品SIM图http://ng1.17img.cn/bbsfiles/images/2011/07/201107271350_307054_1644700_3.jpg再对DBEP标准品通过全扫描并与NIST库比对,几乎与谱库中DBEP质谱图完全符合下图为我进的标准品SCAN与NIST图http://ng1.17img.cn/bbsfiles/images/2011/07/201107271352_307056_1644700_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/07/201107271352_307058_1644700_3.jpg从上图可以看出根本没有205、278这两个的离子的,国标怎么会选呢?请教做过的、有经验的版友,谢谢!
我们做二氯四氟乙氧基苯胺,是用的液相色谱法,可惜没有合适的标准品,
求助,香草扁桃酸和3-甲氧基-3-羟基苯乙二醇衍生物GC-MS分不开,不管是特征离子还是保留时间都相近怎么办,RT,升温程序改善了没用,柱子是DB-5
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
请告诉2-甲基-4-甲氧基-二苯胺的液相色谱分析方法
[color=#444444]测定白油中环己基甲基二甲氧基硅烷的浓度,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]外标法,重复性很差,想请教一下问题在哪里?[/color][color=#444444]色谱条件简单如下:岛津GC-2014,无自动进样器[/color][color=#444444]柱温:100℃;进样口200℃;检测器300℃;[/color][color=#444444]程序升温:100℃(1min)--5℃/min升至150℃(0min)—25℃/min升至200℃(5min)[/color][color=#444444]色谱柱为非极性毛细管柱[/color][color=#444444]分流比:80:1[/color][color=#444444]样品采用正己烷稀释10倍后分析,质量浓度约15%,色谱分析重复性很差,做过此类分析的高手给些建议,谢谢![/color]
3-氨丙基三乙氧基硅烷,分析纯就可以了,谢谢。
[color=#444444]用六氯环三磷腈和乙醇钠合成六乙氧基环三磷腈,重结晶后测其红外,发现在2660处出现一个峰,查了很多资料,也觉得这个产物在此处不该出峰啊 ,请大神来解析解析这个六乙氧基环三磷腈的红外图[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2019/0325/bw196h8429997_1553514396_147.png#opennewwindow[/img][/color]
2-氯乙氧基乙醇 CAS 628-89-7 在气相质谱用什么柱子走的比较好?
[em0808] [em0808] 各位求助一下关于2.5-二甲基-2.5-双(叔丁基过氧基)己烷的测定,性质,反正关于2.5-二甲基-2.5-双(叔丁基过氧基)己烷的任何资料都可以了,在网上都查过了,找到一篇,我先发上来大家看看.有的话麻烦大家分享一下了[~82374~]
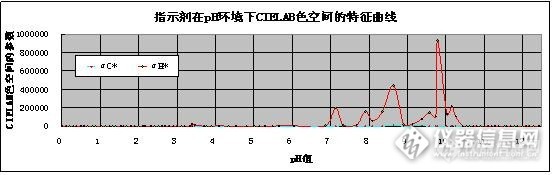
新技术6:乙氧基黄叱精指示剂CIELAB色空间颜色特征摘要:用CIE 1976(L*,a*,b*)色空间方法对乙氧基黄叱精在不同pH环境进行了测量,发现其变色点有8个,与文献记载有较大差异。用于指示的指标a*值、b*值、C*值、△E1、△E1-V可以作为变色的指标,发现在其在高pH环境有新的颜色变化,拟补了传统资料的不足。关键词:乙氧基黄叱精,CIE,色度值,数字化 前言 乙氧基黄叱精(Ethoxychrysoidine Hydrochloride)是常用指示剂,分子式C14H16N4O·HCl,分子量292.77,外观为为深红棕色或黑褐色粉末,在水或乙醇中溶解。pH变色域:pH3.5~ 5.5(红→黄)。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669935_1722582_3.jpghttp://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif图1. 乙氧基黄叱精的化学结构式 采用CIELAB色空间方法研究乙氧基黄叱精指示剂在不同pH溶液中变色现象的文献未见报道。用色空间方法首次测定了乙氧基黄叱精指示剂的L*、a*、b*等色度值参数,与pH值的对应关系,绘制出乙氧基黄叱精指示剂变色的L*a*b*色空间色度学参数与pH值的关系图,为精确描述乙氧基黄叱精指示剂变色特征奠定了基础。1. 实验部分1.1试剂、仪器与测量条件 0.5mol/L H2SO4溶液,0.5 mol/L NaOH溶液,乙氧基黄叱精溶液(取乙氧基黄叱精0.1g,加乙醇溶解、定容至 100ml)。分光光度计,色度测量系统(自研)。1.2 实验内容1.2.1乙氧基黄叱精指示剂溶液的吸收峰 将乙氧基黄叱精指示剂溶液滴入不同pH值的溶液中,在分光光度计测量其吸收峰,见表1、图2。http://ng1.17img.cn/bbsfiles/images/2017/10/2016091607491323_01_1722582_3.jpg图1. 乙氧基黄叱精指示剂在pH环境的吸收曲线 图2显示,乙氧基黄叱精在不同pH值的溶液中的最大吸收峰是不同的。当pH值增加时,吸收峰向短波长方向移动。在pH1、pH2、pH3、pH4、pH5、pH6时,最大吸收峰的波长依次是480nm、480 nm、475 nm、465 nm、450 nm、445 nm。其中在pH3以后至pH6期间变化幅度较大,说明其变色阈值在该范围内激烈变化。pH6以后,即使pH增加,最大吸收峰也是445nm,最大吸收峰保持不变,说明分子结构在pH6以后已经保持稳定,不在变化。表1. 不同PH环境下乙氧基黄叱精指示剂最大吸收峰的变化 PH 波长 1 2 3 4 5 6 7 8 9 10 11 12 445 1.12468 1.43217 1.40751 1.37924 1.23078 0.97832 1.32918 0.93405 1.19237 1.24271 0.94790 1.01212 450 1.21149 1.54126 1.49826 1.43608 1.23844 0.96863 1.31602 0.92405 1.17872 1.22770 0.93672 0.99734 455 1.28742 1.63532 1.57194 1.47602 1.22794 0.94405 1.28232 0.89929 1.14608







 我要推广仪器
我要推广仪器
 下载APP
下载APP