【CEM】环状二硫键桥肽的全自动合成
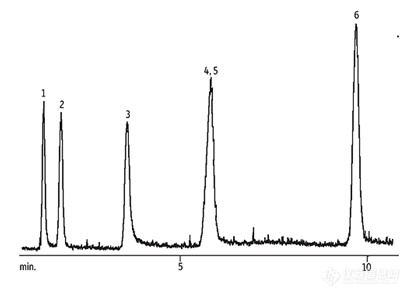
一、引言含有二硫键的环肽代表了一类具有广泛生物功能的化合物,其功能范围从毒素到重要的激素。1二硫键有助于稳定肽的二级结构和构象,这有利于提高蛋白水解稳定性和目标亲和力。2由于它们具有潜在的治疗价值,对合成含二硫键桥连的环肽的兴趣稳步增长。通过使用正交保护的半胱氨酸氨基酸,如Fmoc-(S)-Cys(Mmt)-OH和Fmoc-(S)-Cys(STmp)-OH(图1),可以制备含有二硫键的肽。Cys(Mmt)基团可以使用稀释的三氟乙酉夋(TFA)溶液选择性去保护,而Cys(STmp)基团则使用二硫苏糖醇(DTT)作为还原剂进行正交去保护。去保护后,使用N-氯琥珀酰亚胺(NCS)作为温和氧化剂,可以选择性氧化Cys巯基形成二硫键。3 在这里,我们报告了使用Liberty BlueTM微波肽合成器全自动合成良好纯度的含二硫键桥连的环肽。一个骨形态发生蛋白受体激活素样激酶3 (Alk3)的肽激动剂THR-1234,在3小时内完成合成,纯度为77%。最后,一种含有两个二硫键的锥蜗牛毒素肽(Conotoxin-SI)5在不到4小时内合成,纯度为67%。将微波能量应用于二硫键桥连肽的合成可以实现更高效的偶联,从而快速合成并达到高纯度(CarboMAXTM)。6图1. 左:Fmoc-(S)-Cys(Mmt)-OH;右:Fmoc-(S)-Cys(STmp)-OH二、实验部分HE-SPPS材料和方法:所有肽都是在CEM Liberty Blue&trade 自动微波肽合成器上合成的,使用的是Rink Amide ProTide&trade LL树脂(0.19 mmol/g替换)或Cl-MPA ProTide&trade LL树脂(0.18 mmol/g替换)。使用DMF进行后去保护洗涤,然后采用DIC/Oxyma活化方法进行偶联。肽树脂在CEM Razor® 肽裂解系统上用TFA/TIS/H2O/DODT(92.5/2.5/2.5/2.5)裂解。肽在冷乙酉迷中沉淀,粗品在分析前进行冻干。 分析:粗肽在配备了Acquity UPLC BEH C8柱(1.7 μm, 2.1 x 100 mm)的Waters Acquity UPLC系统上进行分析,该系统装有PDA检测器。UPLC系统连接到Waters 2100单四级杆MS用于结构测定。峰分析是在Waters MassLynx软件上完成的。分离是通过(i)水中的0.05% TFA和(ii)乙腈中0.05% TFA的梯度洗脱进行的。三、结果和讨论A)合成THR-123,CYFDDSSNVLCKKYRS-CO2H选择THR-123(图2)来展示含有C端酸的单一二硫键桥连肽的合成。该肽在10 µ mol规模上使用Cl-MPA ProTide&trade LL树脂(0.18 mmol/g替换)进行合成。第一个氨基酸使用CEM之前报道的氯化物加载循环自动加载。所有其他氨基酸循环使用1分钟/90º C去保护和一次2分钟/90º C与DIC/Oxyma偶联(使用Fmoc-(S)-Cys(Mmt)-OH用于C)。使用2% TFA的DCM溶液进行Cys(Mmt)的去保护。反应在室温下进行1分钟,重复五次。使用25 mM的NCS的DMF溶液实现二硫键的形成。反应在室温下进行15分钟。在Liberty Blue自动微波肽合成器上进行的THR-123的微波增强SPPS产生了77%纯度的目标肽(图3)。图2.THR-123图3. THR-123的UPLC色谱图B) 合成Conotoxin-SI,ICCNPACGPKYSC-NH2选择Conotoxin-SI(图4)来展示含有两个二硫键的环肽的合成。该肽在10 µ mol规模上使用Rink Amide ProTide&trade LL树脂(0.19 mmol/g替换)进行合成。所有氨基酸循环使用1分钟/90º C去保护和一次2分钟/90º C与DIC/Oxyma偶联(使用Fmoc-(S)-Cys(Mmt)-OH用于C;使用Fmoc-(S)-Cys(STmp)-OH用于C)。使用2% TFA的DCM溶液进行Cys(Mmt)的去保护。反应在室温下进行1分钟,重复五次。使用25 mM的NCS的DMF溶液实现二硫键的形成。反应在室温下进行15分钟。使用5% DTT + 0.1 M NMM的DMF溶液进行Cys(STmp)的去保护。反应在室温下进行5分钟,重复三次。最后,使用25 mM NCS的DMF溶液形成第二个二硫键(室温下15分钟)。在Liberty Blue自动微波肽合成器上进行的Conotoxin-SI的微波增强SPPS产生了67%纯度的目标肽(图5)。图4.Conotoxin-S图5. Conotoxin-SI的UPLC色谱图四、结论采用全自动快速合成技术,我们成功高效地完成了含二硫键桥接的环肽的合成,并且实现了较高的纯度。借助CarboMAX&trade 6化学技术,偶联效率得到了显著提升,这不仅极大缩短了合成时间,还确保了产物的高纯度。例如,一个C端带有羧酸的环状二硫键桥接肽——THR-123,在短短不到3小时的时间内就被迅速合成出来,且纯度达到了77%。相比之下,传统的室温合成方法通常需要长达20小时来合成包含两个二硫键的Conotoxin-SI。3而利用微波增强的固相肽合成技术(SPPS),在不到4小时内就制备出了纯度为67%的相应肽。引用1 Góngora-Benítez, M. Tulla-Puche, J. Albericio, F. Chem. Rev. 2014, 114 (2), 901–926.2 Adessi, C. Soto, C. Curr. Med. Chem. 2002, 9 (9), 963–978.3 Postma, T. M. Albericio, F. Org. Lett. 2013, 15 (3), 616–619.4 Sugimoto, H. LeBleu, V. S. Bosukonda, D. Keck, P. Taduri, G. Bechtel, W. Okada, H. Carlson, W. Bey, P. Rusckowski, M. Tampe, B. Tampe, D. Kanasaki, K. Zeisberg, M. Kalluri, R. Kalluri, R. Nat. Med. 2012, 18 (3), 396–404.5 Azam, L. McIntosh, J. M. Acta Pharmacol. Sin. 2009, 30 (6), 771–783.6 CEM Application Note (AP0124) - “CarboMAX - Enhanced Peptide Coupling at Elevated Temperature.”























 我要推广仪器
我要推广仪器
 下载APP
下载APP