RT:10版药典对乙酰氨基酚中的对氨基酚及有关物质、对氯苯乙酰胺检测有个朋友公司开始做这个项目,药典采用的是C8色谱柱朋友想买热电的柱子,不知道有没有同行采用热电的柱子做过?有的话帮忙发一张图谱看看,谢谢!QQ:342832185
CAS:283159-95-5我用C18的柱子,流动相A、B分别是0.05%TFA的纯水、0.05%TFA的乙腈,流动相B的梯度是初始10%,5min升到60%并保持10min,15min后降到10%保持5min,谱图主峰分叉。[img=,690,184]https://ng1.17img.cn/bbsfiles/images/2024/01/202401271531592473_6839_4185279_3.jpg!w690x184.jpg[/img]请问(S)-3-氨基-3-(4-氯苯基)丙酸甲酯是手性化合物吗,需要什么类型色谱柱和流动相条件分析?
3-羟基苯胺和3-氯苯胺缩合反应生成3,3'-二氨基二苯醚,反应具体的条件是什么???文献上只说了加催 化 剂Cucl,吡 啶 (Py),甲 苯和 碳 酸 钾,但没说加多少,所以请高人指点一下,十分感谢啦!!!!!http://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gifhttp://emuch.net/bbs/images/smilies/smile.gif
想请教一下各位网友,我用苯胺衍生二甲氨基甲酰氯,但是随着衍生时间延长,以及苯胺用量,衍生物峰面积一直在增加,做不到反应彻底,有什么解决方法吗
想请教一下各位网友,我用苯胺衍生二甲氨基甲酰氯,但是随着衍生时间延长,以及苯胺用量,衍生物峰面积一直在增加,做不到反应彻底,有什么解决方法吗
[color=#444444]目前本人在做一个培养基中氨基亚砜蛋氨酸(MSX)的检测,考虑到氨基酸类在液相中吸收弱,想到了柱前衍生,查了一些资料用了2,4-二硝基氯苯(CDNB)。[/color][color=#444444]实验条件:1mL 1 mg/mL 的MSX+1 mL Na2CO3 (pH=9.0, 1%)+227 uL 30mg/mL CDNB,85℃暗处反应1h,冷却+0.5 mL 0.1mol/L HCl, 加水定容至10mL,UPLC进样。[/color][color=#444444]色谱条件:[/color][color=#444444]色谱柱:Waters ACQUITY UPLC Peptide BEH C18[/color][color=#444444]吸收波长:360 nm,[/color][color=#444444]流动相:70% NaAC-HAC, 30% ACN[/color][color=#444444]流速:0.2 mL/min[/color][color=#444444]现在出现的问题是:20 min内出的较为明显的峰是CDNB的峰,没有经CDNB衍生后的新的峰出现[/color][color=#444444]我想请教做过类似柱前衍生实验的前辈,是我这个衍生过程有哪里不完善吗?还是色谱条件不对?但这种方法已经很成熟了,文献报道也很多了。还是这个氨基亚砜蛋氨酸不容易被衍生化?[/color]
我现在用气相检测苯甲酰氯,用的SE—54的柱子,什么时间段的峰是氯苯的峰那?
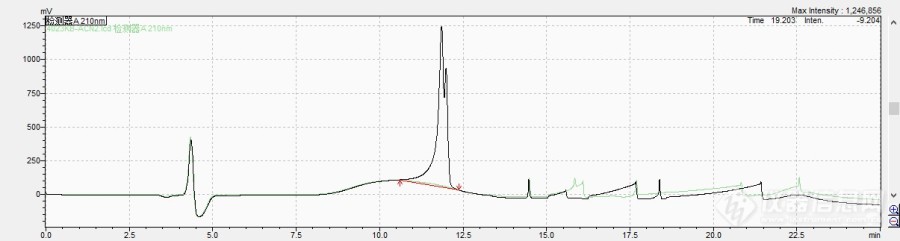
http://simg.instrument.com.cn/bbs/images/brow/emyc1007.gif这是我们实验室几年前做的,拿来参加原创大赛,支持化学药分析版。HPLC法测定小儿氨酚黄那敏片马来酸氯苯那敏含量【处方】 马来酸氯苯那敏 0.5g 对乙酰氨基酚 125g 人工牛黄 5g共制成 1000片1.对照品与供试品马来酸氯苯那敏对照品(中国药品生物制品检定所,批号100047-200305)对乙酰氨基酚对照品(中国药品生物制品检定所,批号100018-200408)小儿氨酚黄那敏片(本公司,批号:20060201、20060801、20060802)小儿氨酚黄那敏片阴性样品(不含马来酸氯苯那敏和对乙酰氨基酚、本公司)2.马来酸氯苯那敏含量测定2.1仪器与试剂2.1.1仪器:岛津LC-10A高效液相色谱仪2.1.2试剂:甲醇、乙腈为色谱纯,磷酸二氢钾、三乙胺、磷酸为分析纯,水为超纯水。2.2测定方法【含量测定】照高效液相色谱法(中国药典2005年版二部附录V D)测定色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;磷酸盐缓冲液(分别取乙腈250ml与0.02mol/L磷酸二氢钾溶液250ml加十二烷基磺酸钠0.68g,振摇使溶解,用磷酸调pH至3.5)-甲醇(10:9)作为流动相,检测波长为215nm,理论塔板数按马来酸氯苯那敏峰计算应不低于3000,马来酸氯苯那敏峰与其他峰的分离度应符合规定。对照品溶液的制备 取马来酸氯苯那敏对照品约20mg,精密称定,置50ml容量瓶中,加甲醇-0.5%醋酸(1:1)混合液适量,振摇使溶解,加上述混合液稀释至刻度,摇匀,精密量取5ml[/fon
有没有人用气相做过对氯苯甲酰氯?气相条件是什么?先谢谢大家!
谁能告诉我“对氯苯乙酰氨”是什么东西,有其他名称吗?
在国标中发现氯苯甲脒与杀虫脒的英文名是一致的,但CAS No.不一样,氯苯甲脒是1970-95-9,杀虫脒是6164-98-3,请问它们是同一种化合物吗?
最近扩项,按照HJ/T 39-1999《 固定污染源排气中氯苯类的测定方法》——无水乙醇洗脱—气相色谱法,对于氯苯类的采样,需要一种很奇特的富集剂。标准原文如下:5.13 富集剂:二乙烯苯与乙基苯乙烯共聚物类多孔高分子小球型载体,比表面积约400㎡/g,颗粒度0.45-0.9mm。事先在脂肪提取器中用无水乙醇(5.1)处理8个小时。晾干后于80℃烘8h,备用采样用的富集柱:于40mm×5mm(内径)的硬质玻璃柱中,填装0.5g富集剂(5.13)并于两段塞少量玻璃棉,或视样品浓度,适当增加柱长度。
谁能告诉我“对氯苯乙酰氨”是什么东西,有其他名称吗?
二乙基二硫代氨基甲酸银除了国药集团还有哪个厂家有?
3,4-二氯苯基异氰酸酯连续化生产工艺研发王绪根 【摘要】:3,4-二氯苯基异氰酸酯(3,4-DCPI)是重要的有机反应中间体,产品主要用于脲类除草剂、农药合成和某些医药合成的中间体。近年由于需求量的增大,急需建立大规模连续化生产装置。本文结合实际情况,通过流程模拟和小试,设计出100吨/年中试生产装置,对中试装置进行安装和调试后,进行了中试,并对异氰酸酯反应动力学进行了研究。 首先,通过查阅文献和实际情况,制定了3,4-二氯苯基异氰酸酯连续化生产的流程:将3,4-二氯苯胺用甲苯溶解,然后将液体光气和3,4-二氯苯胺溶液通入冷反应器,再逐次经过热反应器1、2、3后,流入光化液储罐;确定了生产的主要约束条件和操作条件:系统负荷、光气配比、溶剂配比、系统压力、各反应器的温度和搅拌转速。通过ASPEN和PROⅡ软件模拟,并结合实际情况设计出了3,4-二氯苯基异氰酸酯的100吨/年连续化中试生产装置。 然后,在3,4-二氯苯基异氰酸酯中试生产装置各个设备制造出来后,进行设备安装和调试。在设备调试好之后,先用纯溶剂在拟定条件下进行试车操作。之后,进行了不同操作条件下中试试验。 最后,通过对中试试验结果的整理和分析,可认定3,4-二氯苯基异氰酸酯的连续化生产完全可行。并通过中试找到了3,4-二氯苯基异氰酸酯连续化生产的较优操作条件:在系统压力(表压)10~20KPa;系统负荷为3,4-二氯苯基异氰酸酯150吨/年;冷反应器温度30~40℃,冷反应器搅拌转速750转/分;第一热反应器温度80℃,第一热反应器搅拌转速200转/分;第二热反应器温度90~100℃,第二热反应器转速150转/分;第三热反应器温度120℃,第三热反应器搅拌转速100转/分时,装置运行良好。并进行了异氰酸酯反应动力学的研究。【关键词】:3、4-二氯苯基异氰酸酯 光气 反应器 反应动力学 【学位授予单位】:青岛科技大学
DPD(对氨基-N-N-二乙基苯胺硫酸盐)试剂氧化了,配的溶液都是红的。是因为见光还是夏天温度太高了
也就是说脱色过程不是针对材料类使用的,对于使用在聚酯纤维的分散染料释放的芳香胺比如4-氨基偶氮苯如果不使用氯苯或二甲苯脱色根本就测不出来,我听说国内很多实验室在使用GB/T 17592测试偶氮根本就不用氯苯脱色的过程,这就存在一个风险,像常见的芳香胺比如4-氨基偶氮苯直接就被忽略了,这很容易引起客户投诉或测试纠纷,这种情况怎么办?
急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
最近在看GB5009.19-2008的方法,其中有一个农残 五氯苯基硫醚 从网上找不到CAS号,只能找到甲基五氯苯基硫醚,这两种物质是一样的吗?哪位大侠知道?
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
我是做微生物降解农药的。 我这有个微生物能降解五氯苯甲腈这种物质 生成一个新物质 我想鉴定一下生成的物质。 可是我不知道怎么鉴定 。文名称:五氯苯甲腈中文别名:五氯苯氰; 2,3,4,5,6-五氯苯甲腈; 2,3,4,5,6-五氯苯腈英文名称:pentachloro benzonitrile英文别名:pentachlorocyanobenzene; pentachlorobenzonitrile; 2,3,4,5,6-pentachlorobenzonitrileCAS号:20925-85-3分子式:C7Cl5N分子量:275.3466InChI:InChI=1/C7Cl5N/c8-3-2(1-13)4(9)6(11)7(12)5(3)10分子结构:密度:1.76g/cm沸点:331.7°Cat 760 mmHg闪点:142.8°C蒸汽压:0.000153mmHg at 25°C气质连用可以检测五氯苯甲腈和它的产物吗 不知道用气质的条件 请做分析的高手请教
各位大虾: 最近需要用到邻氯苯基荧光酮这种物质,可是查了很多药品公司都没有货,不知哪位大虾知道广州哪家公司有买或者能否告知具体的合成路线,有关这种物质的任何相关信息都可以! 先表示感谢!
我的一氯苯未响应,三氯苯的响应值大概是二氯苯的10倍,不明白只是差了一个氯离子怎么响应值差这么多啊,同样进的1ul同样的浓度,进1000ug/ML 的氯苯也没响应,自己配的10mg/ML的氯苯才有响应大概峰面积也就100多,不明白响应值会差这么多呢?
国家重申生产者不得在豆芽生产过程中使用6-苄基腺嘌呤、4-氯苯氧乙酸钠、赤霉素等物质,豆芽经营者不得经营含有6-苄基腺嘌呤、4-氯苯氧乙酸钠、赤霉素等物质的豆芽。大家有豆芽中6-苄基腺嘌呤、4-氯苯氧乙酸钠、赤霉素同时检测方法吗?
各位专家: 您好! 我想知道苯胺、氯苯、硝基苯、甲醛、甲醇等对COD的贡献值各为多少,谢谢指教!
谁有:过氧化苯甲酰和N,N-二羟乙基对甲苯氨的标准红外光谱图。
升温条件:40 °C(2min)以10 °C/min到160 °C(2min)。检测器:ECD 温度300°C进样口温度:220 °C进样量:1 μL进样方式:不分流进样色谱柱:DB-FFAP 仪器型号:安捷伦8890[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url] 遇到的问题是:在做甲醇中的4种氯代苯(氯苯、1,4-二氯苯1,2-二氯苯1,2,4-三氯苯)混合时,除氯苯外其余正常出峰,1,4-二氯苯出峰时间大概在7.5min出峰,想求助一下各位前辈,此条件如何优化才能出现氯苯峰。我们最开始是分流进样,后改成不分流进样,1,4-二氯苯前面会出现一个很小很小的峰,但只在标液中出现,按照此峰做曲线标样中的此峰面积带入计算不正确,因为判定此峰可能不是氯苯,很有可能氯苯是没有出峰。求助求助求助,感恩各位!

http://simg.instrument.com.cn/bbs/images/brow/emyc1007.gif发个原创,比较简单,纯支持气相色谱版。氯苯残留溶剂测定帕米膦酸二钠合成过程中使用到了氯苯,属于第二类溶剂。残留限度应不得过0.036%。为控制产品质量,我们参考某品种的氯苯残留测定方法,进行了相关试验。1.帕米膦酸二钠的性质帕米膦酸二钠在水中溶解,在乙醇中不溶,在氢氧化钠试液中易溶。2.方法的选择帕米膦酸二钠不能气化。水溶液呈碱性,若采用溶液直接进样法,容易给色谱系统带来严重污染,因此选用顶空进样法。3.溶剂帕米膦酸二钠在水中溶解,实测取本品约0.1g,加水5ml,振摇,结果溶解良好。因此选用水做溶剂。4.色谱柱首选毛细管柱,柱效高,购置容易。5.测定法取本品0.1g,精密称定,置顶空瓶中,加水5ml使溶解,作为供试品溶液。另取氯苯适量,加水制成每1ml约含氯苯7.2μg的溶液,作为对照品溶液,精密量取对照品溶液5ml,置顶空瓶中,照残留溶剂测定法(附录 VIII P),依法操作。使用固定液为5%苯基-95%二甲基硅氧烷共聚物的毛细管柱,检测器为FID,程序升温,初温为60℃,再以每分钟10℃的速率升温至120℃,维持5分钟。平衡温度80℃,平衡时间30分钟,取顶空1ml,注入气相色谱仪,记录色谱图。供试品色谱图中如显氯苯峰,其峰面积不得过对照品溶液的主峰面积(0.036%)。6.专属性要求:应无干扰峰取帕米膦酸二钠适量,重复精制后,分取两份,各约0.1g,分置顶空瓶中。一瓶精密加入对照品溶液5ml,振摇使溶解后,依法测定。结果色谱图中呈现氯苯的色谱峰。另一瓶精密加水5ml,振摇使溶解后,依法测定,结果色谱图中与氯苯峰保留时间一致的位置上未呈现色
大家都用过哪个厂家的二乙基二硫代氨基甲酸银,价格如何?
我在用二乙基二硫代氨基甲酸银光度法测定砷,有三个问题,第一个为什么要加1ml150g/L的硫酸铜,是不是先抑制[H]的生成,使Zn粒加入后先生成铜,再生成氢,之后产生砷化氢,这样创造时间有利于快速盖上砷反应管?第二个砷发生器的密封性如何做,能否用凡士林,使之不漏气?第三个是反应过程中三氯甲烷的挥发,能否最后补加三氯甲烷,这样是否严重影响结果,其他关于二乙基二硫代氨基甲酸银光度法测定砷的好建议方法,希望能指教,谢谢大家







 我要推广仪器
我要推广仪器
 下载APP
下载APP