氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
[color=#444444]最近在做苯乙酮和1-苯乙醇的化学实验,想用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进行分析,结果发现两个的沸点相差很近,请问这种情况下应该怎么样进行测试,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的操作应该注意什么,苯乙酮的沸点是202.3度,1-苯乙醇的是203.4度,谢谢大家[/color]
哪位同志有对羟基苯乙酮的滴定含量检测方法.我这里有一个方法,但总是滴不好,用甲醇钠溶液进行滴定,用二甲基甲酰胺进行溶解,用麝香草酚蓝作指示剂.目前存在的问题就是终点变化不明显.哪位有用到或碰到类似情况,都来说说吧.
气相色谱仪如何测定苯乙酮的含量,我没学过很想知道,我们厂生产苯乙酮但是不知道怎么用色谱仪做,测定含量
俺想用氨基苯乙酮做显色剂,不知道有人用过吗?还存在邻,间,对的问题,不知道大家用的哪个?
[color=#444444]用液相检测邻氨基苯乙酮时 前处理为什么要在100摄氏度加热5分钟,然后冷却到室温后再检测呢?[/color]
有关苯乙酮的性质如下外观与性状:无色或淡黄色低熔点、低挥发性、有水果香味的固体。熔点(℃):19.7,相对密度(水=1):1.03(20℃),沸点(℃):202.3这么低熔点的物质在室温下(如现在20°左右)基本都是液体了,那么怎么配置30ug/ml的甲醇溶液呢?如何称量?如果用量筒,又怎么计算啊?
测果糖中的二氨基苯乙酮,标准曲线好做吗?为什么我的老是不出峰,且基线不是很稳.紫外220nm,1.0ml/min,柱子C1840度.进样量是300ul很大的,各位有谁做过,给支个招吧谢谢
请问专家能否告诉我对氯苯乙酮的大概分析方法?
间硝基苯乙酮、CAS:121-89-1 质量标准 化学试剂 工业级均可,请大家帮帮忙。
[color=#444444]我在安捷伦[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]上(色谱柱是HP-5)分别进了苯乙酮和α-苯乙醇,两个物质出峰时间差不多,然后我又进了二者的混合物(体积比1:1),结果两峰重叠在一起,我调试了柱温,分流比,都没什么进展。(柱温120,进样温度220,检测器220,分流比60:1),请高手指点一下,是不是色谱柱的问题,还是什么?[/color]
我们需做苯乙酮还原成苯乙醇的反应无副反应,想测其转化率。以前都是用的是面积归一法做,但他想用更精确点的办法解决,请问除了内标法外,是不是可以用测出那两种物质的响应因子来解决?还是用面积归一法就可以了?测响应因子该怎么测?
最近要做这两个物质的检测, 不知道有没有老师在做的给点意见和相关资料,谢谢苯乙酮 98-86-2 2-苯基-2-丙醇 617-94-7
据外媒报道,近日部分德国媒体报道称,珍珠奶茶含有多种致癌成分,严重危害人体健康。此消息一出,德国珍珠奶茶专卖店的生意变得异常惨淡,部分店家甚至面临关门歇业的境地。 如何检测苯乙酮、溴化物及苯乙烯等致癌物?
我们是用WAX柱做苯乙酮,甲醇超声萃取,但有一个样品用GC-MS做有值,质谱、保留时间一致,但是用LC-DAD走样不出峰,标样有出峰,样品加标也有出峰,请问有人知道是什么原因吗?改如何判定?
问题:请教大家 你们乙醇定量是用什么做内标呀?我们现在用苯乙酮 味道太大
新手小白,刚接触GC-MS/MS,我用的是安捷伦7890A-7000B,柱子是DB-5的柱子,走的分别是羟基苯甲酸丁酯和2-氨基苯乙酮的标品,都是1ppm的,溶剂都是丙酮,升温条件50℃——220℃(15℃/min),不分流,然后不出峰。请各位大神多多指导!!!
我们是用WAX柱做苯乙酮,甲醇超声萃取,但有一个样品用GC-MS做有值,质谱、保留时间一致,但是用LC-MS走样不出峰,标样有出峰,样品加标也有出峰,请问有人知道是什么原因吗?改如何判定?
慕尼黑上海分析生化展,迪马科技最新推出了Inspire 苯基系列色谱柱,一直忙未给大家及时普及,今天来说说第一款Inspire PFP ( 五氟代苯基) 是Inspire 液相色谱柱家族新成员,针对分离极性化合物过程中的保留时间和分离度问题而特别设计。Inspire PFP 凭借其优异的选择性,可为极性化合物、复杂天然产物、位置异构体和其它相关化合物在C18 和C8 色谱柱上的分离提供一个替代和补充。Inspire PFP 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式,并具有多种作用机理,因而能够同时分离检测不同极性化合物的混合物,为目前难以解决的复杂极性和亲水性样品的分离分析提供了强有力的工具,可轻松解决其它色谱柱面临的分离难题,为用户实现强极性分析物的优异选择性提供一种更加便捷的途径。同时也为色谱工作者使用简单流动相,避免使用极端pH 条件和准备复杂流动相提供了可能性。Inspire PFP 色谱柱特点• 五氟代苯基硅烷键合在高纯硅胶基质上• 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式• 对极性化合物具有独特的保留能力• 良好的峰形、超高的柱效、分离度和使用寿命• 适用于芳环类化合物或长共轭体系化合物的分离• 优异的批次重现性增强位置异构体分离能力官能团位置的微小差异可以极大的影响分子性能,在许多情况下,传统的C18色谱柱根本无法扑捉到这种细微的差异。然而,Inspire PFP的多功能选择性却可以区分由于分析物内部微小位置变化而导致的分析物的空间位阻变化还是分析物的偶极矩偏移。色谱柱如图所示 规格 150 × 4.6 mm, 5 μm 流动相0.1% 甲酸乙腈溶液:0.1% 甲酸水溶液 = 40:60 流速1.5 mL/min 柱温室温 检测器 UV 254 nm 样品1. 3,4-二甲氧基苯酚 2. 2,6-二甲氧基苯酚3. 3,5-二甲氧基苯酚4. 2,6-二氟苯酚5. 2,4-二氟苯酚6. 2,3-二氟苯酚7. 3,4-二氟苯酚8.3,5-二甲基苯酚9.2,6-二甲基苯酚10.4-氯-3-甲基苯酚11.4-氯-2-甲基苯酚12.3,4-二氯苯酚13.3,5-二氯苯酚http://www.dikma.com.cn/u/image/2016/09/06/1473147613188048.jpg苯氧酸类化合物分子上卤素的加入可以从根本上增强化合物的极性,而极性的变化通常伴随着反相色谱柱在保留时间和分离能力上困难的增加。此时使用InspireTM PFP 是解决保留问题的最有效的方法。InspireTM PFP利用偶极-偶极和氢键作用更好地保留,区分和分离极性卤化化合物。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:0.1% 甲酸水溶液 = 50:50流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 苯氧乙酸2. 邻氯苯氧乙酸3. 对氯苯氧乙酸4. 2,4-二氯苯氧乙酸5. 2,4,5-三氯苯氧乙酸6. 2,4,5-三氯苯氧丙酸http://www.dikma.com.cn/u/image/2016/09/06/1473147817102957.jpg类固醇通过整合偶极-偶极、π-π和氢键机理,InspireTM PFP实现标准反相条件下极性化合物的最佳分离。色谱柱 如图所示 规格 150 × 4.6 mm, 5 μm 流动相甲醇:水 = 60:40 流速1.5 mL/min 柱温室温 检测器UV 254 nm 样品1.泼尼松龙3.地塞米松5.氢化可的松21-乙酸酯7.可的松-21-乙酸酯2.泼尼松4.皮质酮6.11-α羟孕酮8.11-酮孕甾酮http://www.dikma.com.cn/u/image/2016/09/06/1473148006619700.jpg甲基苯乙酮异构体目标分析物上的基团位置变化可以影响化合物的偶极矩,这种变化可以很容易被高电负性的氟原子和其它保留机理察觉,因此InpireTM PFP可以有效地用于分离甲基苯乙酮的位置异构体。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相甲醇:水 = 50:50流速1.0 mL/min柱温室温检测器UV 254 nm样品1. 邻 -甲基苯乙酮2. 对 -甲基苯乙酮3. 间 -甲基苯乙酮http://www.dikma.com.cn/u/image/2016/09/06/1473148212903667.jpg核苷酸和核苷色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相0.1% 甲酸水溶液流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 胞嘧啶2. 5'-CMP3. 5'-UMP4. 5'-GMP5. 尿苷6. 胸腺嘧啶 http://www.dikma.com.cn/u/image/2016/09/06/1473148405914511.jpg抗胃酸药色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:20 mM 磷酸氢二钾(pH 7.0) = 20:80流速1.0 mL/min柱温室温检测器UV 220 nm样品1.法莫替丁2.西咪替丁3.尼扎替丁4.雷尼替丁 http://www.dikma.com.cn/u/image/2016/09/06/1473148597658988.jpg氧化应激标记物色谱柱 如图所示规格 150 × 4.6 mm, 5 μm[/
问一下高手们,氨基苯乙酮的含量高低与温度的高低有一定的比例关系吗?如果液体的话能用活性炭给他除去一些吗?
请教α-甲基苯乙烯,邻甲基苯乙烯,2-甲基苯乙烯,3-甲基苯乙烯,4-甲基苯乙烯,是不同的物质吗?,物质结构
我想了解4-氯-2-三氟甲基苯腈(4-Chloro-2-(trifluoromethyl)benzonitrile,CAS#320-41-2)的理化性质,但在网上只找到沸点109 º C (10 mmHg),是液体还是固体看不出来。因为这个沸点是真空条件下的。那位老师有相关的信息,请告诉我,谢谢!
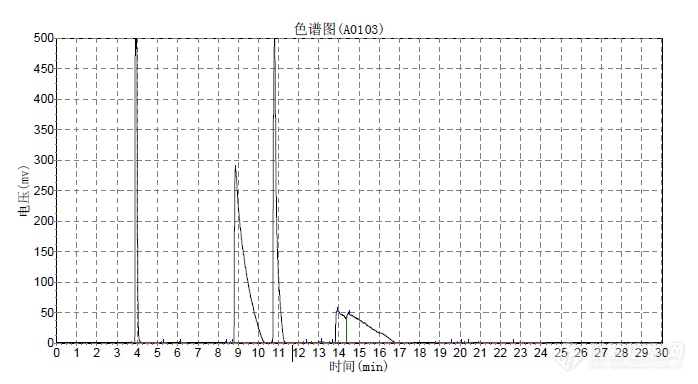
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分离DL-1-苯乙醇,用的CP Chirasil-DEX CB(25m*0.25mm)色谱柱,采用程序升温130度13min,5度每分钟升温到180度,但只有溶剂异丙醇和内标物十二烷的峰正常,苯乙酮峰拖尾,苯乙醇根本分不开R和S。请问是什么原因?[img=,690,379]https://ng1.17img.cn/bbsfiles/images/2019/07/201907101104389918_1404_1750127_3.png!w690x379.jpg[/img]
苯环上3位和5位上各带一个三氟甲基,扫碳谱后非常的杂,如何才能比较准确地找出相对应的峰并计算耦合常数呢?三氟甲基上的碳是否裂分位一个4重峰了,临位的碳也被裂分为双峰了呢?
色谱柱DB23和DB-5都试过,都没有标准品峰进样口温度230,检测器温度250,柱温:60℃,以10℃/min程序升温至120℃,恒温1min,以10℃/min程序升温至200℃,恒温2min,进样量1uL;标准品为1uL苯乙醇溶于2ml乙酸乙酯溶剂峰正常,相同条件下苯乙酮标准品也有峰。为什么1-苯乙醇标准品不出峰???
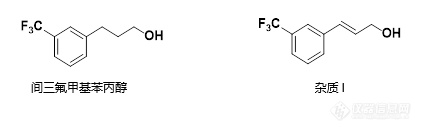
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
请问从哪儿能查到海水中下面的杂质含量测定的方法:甲醇氯化石蜡-52乙二醇二乙二醇二甲基甲酰胺苯乙烯丙酮冰醋酸氯仿甲苯二甲苯1,4-丁二醇苯酚环氧乙烷
有的书上说,三氟甲基在碳谱中可产生1:3:3:1的四重峰,距离比较远。本人做过一次,基本上只得到两个较高峰,两边的小峰有扫不出来的可能吗?而且,三氟甲基对alfa碳有裂分吗?
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
最近做瘦肉精发现福莫特罗和苯乙醇胺a分不开啊,同分异构体,大家有什么办法教教我啊







 我要推广仪器
我要推广仪器
 下载APP
下载APP