适应症脑梗塞,神经内科药理作用本品有改善记忆障碍的作用,能对抗缺氧引起的记忆减退。口服消除半衰期平均22分钟。起效快,作用强,毒性低。用于脑血管病后遗精神行为障碍,可使生活能力提高,记忆再现。无镇静作用。色谱柱信息:XB-C18 5μm,4.6*200mm;PN:ULT 5B18420;SN:210802458色谱条件暂时保密。详见附件,有很大的参考价值的哈,呵呵。。。本文主要探讨茴拉西坦有关物质前处理超声溶解时间不溶对有关物质的影响。1.超声1分钟,进样色谱图:http://ng1.17img.cn/bbsfiles/images/2013/08/201308031518_455752_1621890_3.gif2.样品超声10分钟处理后进样色谱图:http://ng1.17img.cn/bbsfiles/images/2013/08/201308031519_455753_1621890_3.gif3.样品超声10分钟处理后进样色谱图:http://ng1.17img.cn/bbsfiles/images/2013/08/201308031519_455754_1621890_3.gif4.不同超声时间色谱图对比。.http://ng1.17img.cn/bbsfiles/images/2013/08/201308031519_455755_1621890_3.gif5.结论:初步估计是由于超声时间较长,使其辅料溶解出来了,或是由于超声室水温身高(冰浴试验过),10分钟主峰后产生的杂质在15分钟后消失了,此杂质不稳定?抑或是此杂质再降解为其他杂质,从10分钟和15分钟色谱图中色谱峰面积之和推出,这样在物料上平衡才说的过去,头痛,具体情况没有进行再详细的研究。或是本品口服消除半衰期平均22分钟,表明本品不稳定,超声不同时间对该品稳定性有极大的影响。
求助各位大侠,对齐拉西酮怎么分析?用液相色谱的话什么柱子及条件比较好?非常重要,请各位指点http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
急急急!!!实验室检测的哌拉西林酸在水中的溶解度不合格,哪位亲有相关的文献资料送上,或者说说与那些因素有关,不胜感激
求助下起始物料4-溴甲基喹啉酮的有关物质检查方法
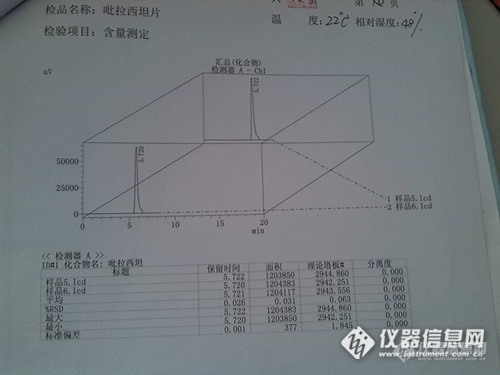
菜鸟———征服吡拉西坦片含量测定 本人是一个药品检验的新手,世称菜鸟。这是第一次做化学药品检验,在一位多年从事药品检验的师傅指导下,做了吡拉西坦片含量测定的检验。如有不妥之处请各位老师批评指正。 吡拉西坦片是化学药的一种,为白色或类白色片。其含量测定按照《中国药典二部2010版》的规定检测。正文P337。 含量测定的具体程序:取本品20片,精密称定,研细,精密称取适量(约相当于吡拉西坦0.1g),置100ml量瓶中,加流动相适量,振摇使吡拉西坦溶解,用流动相稀释至刻度,摇匀,滤过,精密量取滤液5ml,置50ml量瓶中用流动相稀释至刻度,摇匀,精密量取10ul注入液相色谱仪,记录色谱图,另取吡拉西坦对照品,同法测定。按外标法以峰面积计算,既得。色谱条件与系统适用性检查: 用十八烷基硅烷键合硅胶为填充剂;以甲醇:水(10:90)为流动相,检测波长为210nm。理论板数按吡拉西坦峰计算不低于2000。 仪器设备为岛津液相色谱,配有紫外检测器,自动进样器。这是本次检品吡拉西坦片。http://ng1.17img.cn/bbsfiles/images/2014/08/201408291604_512123_2764104_3.jpg对照品谱图。http://ng1.17img.cn/bbsfiles/images/2014/08/201408291607_512124_2764104_3.jpg检品谱图。检品做了两个平行,分别进了两针。http://ng1.17img.cn/bbsfiles/images/2014/08/201408291615_512131_2764104_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/08/201408291613_512129_2764104_3.jpg测得本检品含量为:97.4%。相对平均偏差:0.4%。符合药典规定。
我用岛津10A测头孢哌酮他唑巴坦钠有关物质,为什么用相同的流动相测定,两个主峰的出峰时间(9和21左右)跟文献报道(2和8左右)差很大?而且他唑巴坦还分离不好,分离不完全,大家有谁做过相关的内容么,希望指导一下。
[em17] 本人刚刚入行不久,遇到如下一个难解问题: 苯丙酮酸钙 的 HPLC 有关物质检测方法, 请各位高手都来指点一下。 关于这个试验我试过阳离子交换柱,流动相是0.0125mol/L硫酸,检测波长205nm ,可是峰形不正,严重拖尾,各位朋友谁知道更好的检验方法啊,希望能点拨一下!! 谢谢大家!
请问各位大侠,开发[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]方法检测有关物质主要应注意什么呢?例如不能让主成分分解等,怎么知道主成分没有被分解呢,还有就是怎样能证明[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]方法可用于某种物质有关物质的检测呢?(该物质没有紫外吸收)
各位高手好! 刚学做手性不久,这两天做奥拉西坦,文献用的是大赛璐OC柱,流动相是正己烷与乙醇,我这边没有OC柱。用过正相AD,OD,OJ,IC柱,流动相用过乙醇,异丙醇,正己烷;反相AD,OD,OJ,SCA,chirobiotic T柱,流动相用过乙腈,甲醇,水;添加剂还没有试过,都没有做出来; 有没有哪位大侠做过或是相似结构的,OC柱能用我有的这些柱子配哪种流动相代替呢,请给小女子指点一二,不甚感激!!
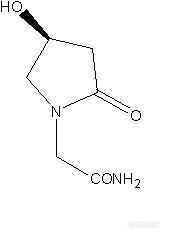
各位高手好! 刚学做手性不久,这两天做奥拉西坦,文献用的是大赛璐OC柱,流动相是正己烷与乙醇,我这边没有OC柱。用过正相AD,OD,OJ,IC柱,流动相用过乙醇,异丙醇,正己烷;反相AD,OD,OJ,SCA,chirobiotic T柱,流动相用过乙腈,甲醇,水;添加剂还没有试过,都没有做出来; 有没有哪位大侠做过或是相似结构的,OC柱能用我有的这些柱子配哪种流动相代替呢,请给小女子指点一二,不甚感激!!http://ng1.17img.cn/bbsfiles/images/2012/04/201204082229_360092_2327592_3.jpg
有关物质问题大家好! 我最近检验一个药品的有关物质,叫“注射用细辛脑”,时常失败,有时会成功。现在查不出原因。如果做有关物质,其影响因素有哪些?有人做过否?
求助各位:在单位用waters2695液相做左乙拉西坦,流动相:甲醇:ph=4.5的纯水=3%:97%;由于紫外响应在210nm,感觉出来的谱图基线超不平,杂质超多,血浆里杂质也多的不得了。。。。可是我的目标峰面积很小,峰型差还拖尾,感觉就是做不下去。可是领导坚持让用液相做,听崩溃的,有没有大虾有办法呢?
建立一种灵敏的检测人尿和血浆中劳拉西泮的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法。酶水解的尿和血浆样品在pH=9用乙醚提取,提取物用MTBDMS衍生化,衍生物用HP-5毛细柱进行色谱分离,用电子捕获检测器检测。尿和血浆中提取率分别为844%和813%,检测限分别为42ng/mL和24ng/mL。方法已用于口服2mg劳拉西泮人的尿和血浆分析,分析结果表明本方法适合于麻醉抢劫案中药物分析。本方法已与非衍生化、TMS衍生化[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法比较,本方法是最灵敏的方法。
木糖醇有关物质[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],用DB1701毛细管柱,半乳糖醇和山梨醇分离度达不到2.0,请问有谁做过这个项目吗?
阿奇霉素的有关物质,系统适用大家都怎么做的,买到适用性的对照品了吗
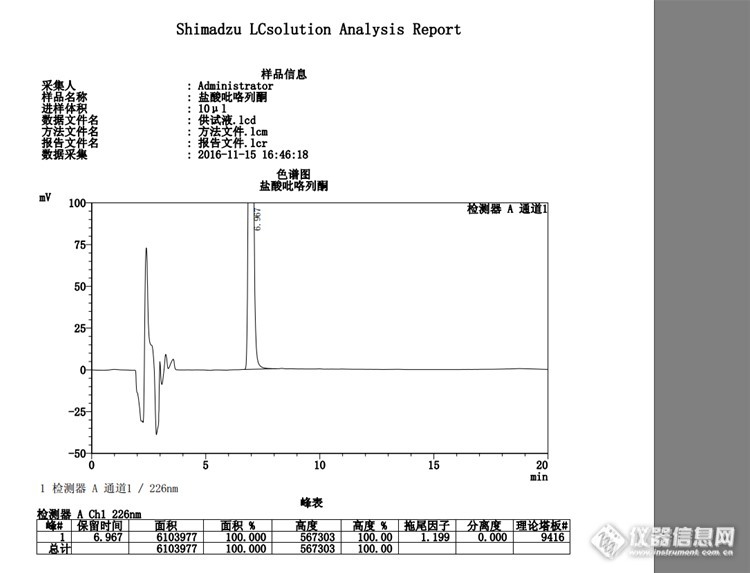
迎接2017年,原创1-Welchrom® C18测定盐酸比格列酮有关物质色谱柱sn:W13211841,pn:00310-02041(5μm,150mm×4.6mm)色谱条件自拟:流动相为乙腈-0.1M醋酸铵溶液(50:50),用醋酸调节pH值至5.0,检测波长为226nm,进样量为10μl,分析时间约为20分钟。操作:取本品适量置20ml容量瓶中,加流动相适量使溶解并稀释制成每1ml约含盐酸比格列酮0.5mg的溶液作为供试液,精密量取1ml,置100ml量瓶中,加流动相稀释至刻度,摇匀,作为对照溶液。本品空白溶剂为流动相。盐酸比格列酮简介药品名称: 盐酸吡格列酮别名:安可妥;盐酸匹格列酮;盐酸吡格列酮;盐酸皮格列酮;匹格列酮盐酸盐英文: Pioglitazone Hydrochloride;ACTOS中文化学名(±)5-苄基]-2,4-噻烷二酮盐酸盐剂型: 原料药性质:外观 白色晶体粉末熔程 193-194℃可溶性:在甲醇中溶解,在乙醇、氯仿中微溶分子式:C19H20N2O3S·HCl分子量:392.90CAS号:112529-15-4用途:新一代二型糖尿病治疗药,副作用小。本品主要成分是盐酸吡格列酮,其化学名称为(±)-5--苯甲基]噻唑烷-2,4-二酮单盐酸盐结构式:[url=http://p1.qhmsg.com/t01f3c03f21df4dc8b0.jpg][img]http://p1.qhmsg.com/dr/220__/t01f3c03f21df4dc8b0.jpg[/img]分子式:C19H20N203S·HCl分子量:392.90本品主要检测有关物质:其典型色谱图如下:空白溶剂:[img=,690,597]http://ng1.17img.cn/bbsfiles/images/2017/01/201701012253_01_1621890_3.png[/img]对照溶液:[img=,690,640]http://ng1.17img.cn/bbsfiles/images/2017/01/201701012254_01_1621890_3.png[/img]供试液:[img=,690,527]http://ng1.17img.cn/bbsfiles/images/2017/01/201701012256_01_1621890_3.png[/img]总结:现在该药品正在处于摸索阶段,结果显示本品较为“干净”,到底干净是否,要等组织长期的调查,我们相信群众的眼睛的,目前色谱数据较为理想,月旭的色谱柱的柱效较高均达到了9000,拖尾因子均小于1.3。一般我们在进行新的项目摸索方法的时候也在摸索色谱柱,初步确认该品种用月旭这款色谱柱较为理想。
客户选型时送的样品,伍丰工程师整理的数据,请大家多提宝贵意见!

最近在做药用辅料(月桂氮卓酮)的有关物质测定,条件和结果如下,(分流比为25:1,且14分钟的大峰是有效成分峰,其余的小峰是杂峰)想请教一个问题,做有关物质测定能否设分流比,我的想法是设了分流比,一些含量低的杂质会测不出来,影响结果准确性。但是不设分流比,又会出现杂峰很多,一是有些分不开,而且溶剂峰很大,拖尾,积分非常不好处理。大家怎么看,第二张图是设了分流比为25:1的[img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905051558063170_8144_3480890_3.jpg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905051606410323_4695_3480890_3.jpg[/img]
有关物质研究一直是药物药学研究和评价中的重点和难点。随着我们对有关物质认识的不断深入,对有关物质的技术要求也在不断提高,其中对有关物质进行定性(鉴定)研究已经成为有关物质研究技术要求的重要组成部分,受到广泛关注。但目前有关物质的定性研究仍然是国内有关物质研究中的薄弱环节,研究水平与国外先进国家相比有较大的差距,也限制了我国药品研究水平和药品质量提高,需要引起国内研发者的充分重视,主动进行深入研究,提高研究水平。 对药物中的有关物质进行定性研究具有重要意义,通过对有关物质的定性研究,我们可以获得有关物质的结构信息,分析其形成过程,合成时可设法避免该杂质的形成,或经纯化使之降至可接受的水平。另外,还可以通过检索毒性物质数据库获知该杂质的毒性数据,为其限度的确定提供有力的依据。同时,定性研究也是分析方法确定的重要参考,对贮藏条件的确定也有指导意义。如果不了解有关物质的结构,后续的研究将无法继续进行。 对药物中的有关物质进行定性分析也是我国药品研发与国际接轨的需要。按照ICH Q3A/Q3B指导原则以及2005年SFDA《化学药物杂质研究技术指导原则》的要求,对药物中超过鉴定限度的有关物质皆应明确其来源,并推测可能的结构。我国的药物研发要想与国际接轨,就必须在有关物质研究的完整性和规范性上符合相关的要求。 按照研究方法的不同,有关物质的定性研究可以分为理论推导法、直接测定法和间接测定法等,在实际的应用中,这几种方法常常结合使用,相互印证。一般而言,由于有关物质的定性研究不能像原料药的结构确证一样提供全面的信息,因而需要尽可能采用多种方法(直接测定法例外),提供尽可能多的信息和证据,否则,有可能得到错误的结论。下面对几种方法进行分别说明:
以下两个物质分别为占吨酮和占吨酸,为2010药典溴丙胺太林的有关物质,现在需要采用TLC将两物质分开,我根据2005药典溴丙胺太林有关物质的方法:二氯乙烷:甲醇:水:甲酸=56:24:1:1,两者均在展开剂的前沿,且无法分离,基本是重合,我试过将展开剂的极性提高,提高了甲醇的比例,从50:50提高到5:75,虽然Rf有所降低(从1降至0.8),但两者仍然无法分离,也试了乙酸乙酯以及乙酸乙酯:甲醇=10:5,均无法分开大家能否给点意见,采用什么方法可以分开呢[img]http://ng1.17img.cn/bbsfiles/images/2010/08/201008112151_235990_1919795_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/08/201008112152_235992_1919795_3.gif[/img]
阿奇霉素有关物质分析方法对色谱填料的要求目前2005版中国药典对阿奇霉素相关物质的测定采用薄层色谱法(TLC),对含量的测定是采用微生物检定法,方法的专属性不好。USP以及欧洲药典现行分析方法是采用液相色谱法。
想求一张用WAX(30m*3.2mm,0.5um)做的丙二醇有关物质对照品溶液的色谱图,希望能附上升温程序,急需
有些药品需要做两个有关物质,这两个有关物质有什么不同?
关于液相的有关物质:2010版本药典增加了很多产品的有关物质例如阿奇霉素的有关,其中有阿奇霉素B(相对保留时间1.53),阿奇霉素Gx(0.61),二氢高红霉素(0.4),红霉素A偕亚胺醚(0.2)各种杂质标准中只提到这些杂质和阿奇霉素的相对保留时间,如果附近有几个杂质峰,但是相对保留时间都不是很准确,如何去判断哪个是我们要的杂质呢?
在做有关物质方法验证时需要测定检测限。依据中国药典2005版2部附录药品质量标准分析方法验证指导原则,检测限可以用信噪比法测定,即把已知低浓度试样测出的信号与空白样品测出的信号进行比较,算出能被可靠地检测出的最低 浓度或量。一般以信噪比为3 :1 或2 :1 时相应的浓度或注入仪器的量确定检测限。在杂质已知的情况下,可以通过配置杂质溶液测定其最低 浓度。但在杂质未知的情况下,怎样确定有关物质的最低 检测浓度?大部分文献中,有关物质的最低 检测浓度采用供试样品溶液稀释或对照品溶液稀释进行测定,这样测定出的是主药的最低检测浓度,而不是其有关物质的最低检测浓度。
给出原料药和制剂的有关物质限度,怎样判断其是否合理?
作有关物质的实验时,要求做降解物质,即酸破坏、碱破坏、高温、强氧化有什么依据么?还有“调节仪器灵敏度使主成分高为满量程的10%--20%”是调节检测器的灵敏度吧?可是卖仪器的人说不可以随便调的,还有检出限的问题,直接稀释已知浓度的样品可以么?因为说信噪比也不可以调的,尤其对于老的仪器,新手求助,谢谢
作为一个新药,对其纯度的检查是保证安全有效的重要指标之一,而纯度检查的内容,根据各个药物的性质和特点有些不同,但基本上均要涉及各自的“有关物质”检查研究。关于有关物质的检查,在国际上已早为“人用药品注册技术规范协调会(ICH)”所关注,国内在新药的有关文件中也提出了供参阅的一些具体要求,并逐渐引起研制新药者的重视。 由于一个新药从合成原料药到制备有关的制剂,再经贮藏、运输、使用,要经历一段较为复杂和漫长的过程,在此期间,每一过程都有可能产生有关的物质,如生产中可能带入起始原料、试剂、中间体、副产物和异构体等;在贮藏和运输过程中,可能产生降解产物、聚合物或晶型转变等特殊杂质。为保证药物的安全有效,同时也要考虑到生产实际情况,因此,国内外对药物的研究,可允许含有一定限量的无害或低毒性的有关物质,但对毒性较大,能危害人体健康的、无效的或能影响药物稳定性的有关物质则必须严格控制。 因有关物质的量微小,故检测方法至关重要,必须选择专属性强、灵敏度高、重复性好的方法。目前,首选的是色谱法,可根据新药的具体品种及其有关物质的性质采用TLC,HPLC及GC等,有的还应采用其他有关的色谱或波谱法。对于一、二类新药更应重视,必要时可采用HPLC/UV二极管阵列、HPLC/MS或GC/MS等联机技术,对有关物质进行定性定量分析。 现将有关物质检查中常见到的一些问题讨论如下。1 未提供研究方法的专属性 申报资料中常见到有关物质检查的结论是“未检出有关物质”,甚至在稳定性各影响因素考察中,样品颜色已明显变化,但其结果仍是“未检出降解产物”,因而提供的TLC图上只有1个斑点,HPLC图上只有1个主药的峰。对于此类情况,应考虑方法的专属性,如采用的是HPLC,则在方法研究中可按中国药典附录的要求,进行系统适用性试验,考察方法的专属性,具体方法有:①如有关物质为已知的某中间体、副产物或降解产物,则可在原料药中加入该杂质适量进行试验,以证明能达到分离;②如有关物质为未知,可用含有杂质的粗品进行试验,以证明能达到分离;③将精制品经强光、高温、高湿、氧化剂或酸碱等分别处理,使样品光解、分解、氧化或水解后进行试验,以证明能分离出杂质。经上述方法试验能分离出杂质,即证明采用的方法具有一定的专属性。2 未提供研究方法的灵敏度 申报资料中,有的把试验的最低浓度作为灵敏度,甚至有的只有文字叙述而无任何数据和图谱,而未提供详细方法学研究资料和图谱来证明该最低浓度为最低检出量。因此,提供的灵敏度缺乏试验依据,不能反映所采用方法的真实检出灵敏度。对此,研究者在研究的方法具有专属性后,应考虑方法的灵敏度。如采用HPLC分析时,一般使最低检出量相当于基线噪音的3倍峰高时注入的样品量;如采用TLC分析时,应将样品配制成一系列的不同浓度,分别点样、展开、显色,以确定灵敏度和最低检出量。3 未提供定性和定量的依据 在申报资料中,除极少数用已知杂质作对照,对样品中有关物质进行定性、定量外,多数强调了制备杂质对照品的困难。因此,对有关物质的定性和定量是用主成分自身对照法或面积归一化法,但因未提供所检测的杂质在设置的检出波长处的响应因子,而不同杂质的的响应因子有可能不同,且差别较大,至使虽有定性和定量结果,但可靠性较差。因此,有关物质定性、定量的最佳方法是采用已知对照品的方法,使所提供的定性和定量有充分依据。4 未提供有关物质的来源及其性质 申报新药中的有关物质,绝大多数为未知,即使其有关物质含量大于2%或一类创新药,也常把在HPLC主峰以外的各杂质峰统归为未知的杂质,而未研究其杂质的来源、性质及其确切的含量。为保证新药应有的纯度,特别是一类新药,对有关物质检查应严格要求,对其主要杂质要确证化学结构,对主要杂质和其他杂质要分别定出限量。5 色谱图记录时间太短或进样量过小 在资料中,有不少HPLC图谱不符合要求,表现在主峰的保留时间记录刚完毕,即停止记录,致使一些比主成分峰保留时间大的有关物质不能检出或进样量过小,杂质检不出。由于有的药物与其有关物质分子结构的极性相差较大,因此,在HPLC分析中,有关物质的保留时间可能在主成分峰之后出现,故记录时间应比主成分峰的保留时间延长2~3倍,并应考虑样品配制浓度或进样量致使主成分峰为一矩形峰,以确保样品中的有关物质能全部检出。6 提供的色谱图不合要求 资料中常见提供的色谱图失真,例如TLC分析提供的是手工绘制的正圆形示意图,或虽是图谱的照片,但无样品点样点,无溶剂前沿,斑点未标示Rf值,样品无批号或文字说明等;HPLC分析中亦有用手工描绘图谱的,有的原图缩得过小以至图上数据模糊不清,或提供的图谱无样品批号,无保留时间,无文字说明,或各峰积分值与列表数据不相吻合等;在稳定性考察降解产物中,上述情况更为严重,致使无法审查。因此,研究者应认真参考新药审批的有关技术指导原则进行试验,提供符合要求的色谱图。7 资料前后缺乏相关性 在有关物质检查研究中,有的用HPLC进行了较深入的方法学研究,以已知中间体或杂质作对照,证明方法较好,专属性较强,但在制订质量标准研究中的图谱却未出现相同的杂峰,而是其他杂质峰,致使样品研究中的有关物质与制订标准中的有关物质不相关,且在资料中对此无说明。对此情况,应进一步深入研究,弄清是产品工艺不稳定带入的其他有关物质,还是产品本身不稳定的降解产物或残留有机溶剂所致,使研究的结果与制订标准的结果一致。8 未说明有关物质的个数及其总量 资料除极少数研究了有关物质的个数和总量,并放入质量标准外,大多数对有关物质的数和量未做精密研究,对有关物质只是笼统的订为不得大于百分之多少,这就难以保证产品的有关物质是否稳定在规定的限度范围内。因此,在研究并确定了有关物质数和量的基础上,应对若干批工艺稳定后的产品进行分析,积累数据,对有关物质订出个数,并对主要有关物质、未知及总的有关物质分别订出限量,以保证产品质量。
请问怎样着手进行一个药物的有关物质研究?我研究的药物虽属有机物,但无紫外吸收。进行其有关物质研究需考虑哪些因素?需查阅哪些方面的资料?谢谢有经验的人指点帮忙。谢谢!
含量的高低会不会影响有关物质的高低,含量高,有关物质就会低,含量与有关物质之间的关系,有没有大佬解释一下,萌新化验员







 我要推广仪器
我要推广仪器
 下载APP
下载APP