有没有人检测过N-甲基哌啶
2-氯乙基氯甲基醚,CAS:1462-33-5,英文名称叫Chloroethyl chloromethylether,但是在网上和化工大辞典上都找不到它的相关物理化学性质,愁啊
三丙酮胺沸点205四甲基哌啶胺沸点188-189[img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037318102_4160_3963991_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037319030_2268_3963991_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037317834_4061_3963991_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106151037318341_2757_3963991_3.png[/img]
我公司现生产的一种产品,四甲基哌啶醇是精制而成包装时需对进行挥发份进行测定,但该产品易于升华,故现在还没有一个合适的方法,测定溶剂是否已烘于。
腐乳中二甲基黄和二乙基黄的测定解决方案二甲基黄和二乙基黄,属于工业染剂,主要用于石蜡、塑胶、印刷油墨、石油和肥皂等的着色,具有致癌性。因其不属于食品添加剂范畴,从未列入台湾监控部门的常规检查项目,被当地不法商人利用至今。目前,关于该类物质的测定方法几乎没有报道。方法优势:迪马科技建立固相萃取-超高效液相色谱串联质谱法同时检测腐乳中二甲基黄和二乙基黄,本方案具有以乙腈为提取液,采用ProElut DYC固相萃取柱净化样品,通过UPLC- MS/MS检测;前处理步骤简单、净化效果好、回收率高、基质效应小优点;保证实验结果准确性、重现性。方法检出限0.03 μg/kg,定量限为0.1 μg/kg;适用于各省市出入境、质检、疾控、食品药品检验所、第三方检测机构、食品检测机构等。专用柱优势:ProElut DMY 柱由2种吸附剂按照一定的比例分层填装而成,采用不同作用机理去除杂质,同时对二甲基黄和二乙基黄没有不可逆吸附,保证了样品的净化效果及回收率;本产品是商品化的成品柱,不用手工填装,吸附剂稳定性好,不受外界环境因素影响,保证实验结果的重现性和准确性;过柱过程操作步骤简单,节省时间,提高了工作效率以下为详细解决方案,敬请参考!腐乳中二甲基黄和二乙基黄的测定1、适用范围 适用于腐乳中二甲基黄和二乙基黄的检测,二甲基黄的方法检出限是0.03 μg/kg,二乙基黄的方法检出限是0.04 μg/kg,定量限是0.1 μg/kg。2、提取取1.0 g样品,加1.0 g氯化钠与5 mL乙腈,涡旋混匀,6000 rpm下离心2 min,精密量取2.5 mL上清液待净化。3、净化——ProElut DMY 3 mL(Cat#:65914)a活 化:3 mL乙腈活化; b上 样:c淋 洗:加入待净化液,弃去流出液;加入3 mL乙腈,弃去流出液;d洗 脱:加入4 mL10%氨水甲醇,收集流出液;d重新溶解:将流出液在50 ℃下氮吹至干,用乙腈定容至1 mL,过0.22 μm微孔滤膜,供LC-MS分析。4、分析条件4.1 UPLC 条件:色谱柱:Endeavorsil C18,100 × 2.1 mm,1.8 μm (Cat.# 87003)流 速:0.2 mL/min进样量:5 μL柱 温:35 ℃流动相: A:0.1%甲酸水 B:乙腈梯度设置时间/Min.055.510A(%)20102020B(%)809080804.2 质谱条件:电离模式:ESI扫描方式:正离子扫描检测方式:多反应监测电喷雾电压:5500 V雾化气压力:50 psi辅助气压力:50 psi气帘气压力:20 psi离子源温度:500 ℃定性离子对、定量离子对、碰撞气能量及去簇电压见下表药物名称定性离子对定量离子对碰撞气能量/eV去簇电压/ V(m/z)(m/z)(母离子/子离子)(母离子/子离子)二甲基黄226.2/77.0226.2/77.02877226.2/134.128二乙基黄254.2/120.2254.2/120.23473254.2/148.126254.2/134.1335、添加回收结果腐乳中二甲基黄和二乙基黄的LC-MS检测添加回收结果分析物基质添加水平(μg/kg)回收率(%)二甲基黄黄色腐乳1.095.4二乙基黄1.098.6二甲基黄红色腐乳1.090.5二乙基黄1.092.3http://www.dikma.com.cn/u/image/2016/01/29/1454056478555325.jpg二甲基黄标准多反应监测色谱图http://www.dikma.com.cn/u/image/2016/01/29/1454056486222259.jpg二乙基黄标准多反应监测色谱图http://www.dikma.com.cn/u/image/2016/01/29/1454056492122223.jpg添加水平为1.0 μg/kg黄色腐乳中二甲基黄和二乙基黄检测的多反应监测色谱图[/alig
有哪位朋友做过哌啶基哌啶的分析,能否跟我联系下,有一些问题想请教谢谢
(求助)3-乙基-5-(2-羟乙基)-4-甲基噻唑溴 的分子式以及供应厂家。
最近准备参照药典开展LC-ICPMS联机测试总汞、甲基汞和乙基汞的实验。之前没有做过联用,据悉甲基汞和乙基汞都是剧毒物质,请问各位有相关经验的版友,开展此项实验需要做好哪些个人防护措施?做到什么级别?在你们的实验室里都是如何操作的?先谢过各位大虾!
[color=#444444]我用HP-5的毛细管柱和ECD检测器检测氯化甲基汞和氯化乙基汞,仅在1.6min左右有一强峰,难道是两种物质没分开吗?[/color][color=#444444]是柱子没选好还是条件没设定好呢?恳请大家帮忙,烷基汞用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]该如何检测呢。[/color]
2010年版药典氟哌啶醇片红外鉴别方法是:取本品粉末适量(相当氟哌啶醇约50mg),用20ml三氯甲烷分次研磨使溶解,过滤,取滤液水浴蒸干,残渣减压干燥后,压片,与标准图谱比较. 因为氟哌啶醇易溶于三氯甲烷,我在做的过程中就没有剥片;另氟哌啶醇对光 热具不稳定性,滤液采用60度水浴蒸至近干,采用药典原料项下的方法干燥:即60度减压干燥,约5小时,但取出压片时发现它的性状为半固体,压出的片透明度不好,还粘模具,没法做! 会不会是辅料有影响呢?想请教一下,如何改进我的试验,压制一个好的片子?
水产品中甲基汞、乙基汞的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MS法测定 浙江 WUPINGGU 由于环境日益恶化造成重金属污染问题特别突出,如广东珠三角“镉”污染、浙江等地的含“铅”茶叶等等,重金属通过食物链从水开始向食物迁移,并一级一级浓缩转化,其中汞是重金属中对环境及食物影响较大的一个元素,特别是汞经过生物转化从无机态生成甲基汞、乙基汞等有机产物,使其毒性大大增加,国内外对水产品中总汞残留检测同时要求检测甲基汞含量,我国水产品中甲基汞限量值为0.2mg/kg。目前我国GB/T5009推荐的甲基汞测定方法为巯基棉吸附[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/ECD方法测定,但本人在检测过程发现甲基汞峰形不好,灵敏度偏低,巯基棉吸附回收低等缺点(不排除本人实验有误,没有掌握好),其他文献报道的方法主要有液相色谱-原子荧光联用、液相色谱-等离子发射光谱质谱联用、液相色谱质谱等方法,但上述3种方法均要求特定的联用仪器,目前方法推广有局限性,而[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱虽然也是仪器联用,但技术已经适当成熟,而且仪器的普及已经相当广泛,因此本研究结合巯基乙酸对样品中的甲基汞、乙基汞的特异性吸附,然后将甲基汞、乙基汞经过乙基化衍生来改变脱尾的峰形,最后采用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MS对甲基汞、乙基汞进行定性定量分析。1、仪器 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MS DB1701MS柱,30m*0.25mm*0.25um,35度,2min—10度/min-150度。 监测离子:甲基汞 202、215、217、246;乙基汞202、231、258、260。(其他省略)。2、样品提取:称取样品0.5g,加1mol/L KOH甲醇溶液5mL,在100度下水解1小时,取出冷却,离心取上清夜加1mol/LEDTA-2Na溶液0.3mL,加水2mL,用2mol/L盐酸溶液调节pH至5.0,然后加水至10mL,5000rpm离心5min,取上清夜备用。3、样品净化:取巯基乙酸键合固相萃取柱(250mg/6mL),先用5mL淋洗柱子,然后将上述上清液过柱,再用5ml水淋洗除杂,真空抽干水分,加1mL苯洗脱(抽干苯,以防止损失,注体积不会变化多少!)。4、样品衍生:在苯溶液中加入2mol/L醋酸/醋酸钠缓冲液0.1mL,0.1mL四乙基硼化钠溶液,混匀反应15min,然后加入2mL2mol/L NaOH溶液,混匀除去巯基乙酸,5000rpm离心5min,取上层苯溶液进行[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MS分析。结果与讨论(待续)化了1.5小时草草的写了一下近期“水产品中甲基汞、乙基汞的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MS法测定”,希望对大家有所帮助。由于图谱在单位,上班后附上。[em09510]
[size=14px] [/size] [size=14px]丹参酮是中药丹参的功效物质,丹参酮I(Tanshinone I,Tan I)是丹参酮的一个亚类,具有芳香二萜醌的结构,具有抗菌和抗炎活性。然而,其过高的亲脂性,效价较弱,溶解度低、不稳定等特质,极大地限制了丹参酮I的应用。因此,建立有效的化学演化机制,开发更有效的丹参酮Ⅰ衍生物具有重要意义。哌啶是一种重要的饱和杂环支架,是美国FDA批准的药物中最常用的氮杂环,具有良好的药理特性。该团队将丹参酮Ⅰ和哌啶的骨架杂交,得到了一类新型有效的NLRP3炎症小体抑制剂。2023年2月14日,浙江大学药学院的崔孙良和王毅团队在J Med Chem(IF=7.3)上发表题为“Scaffold Hybrid of the Natural Product Tanshinone I with Piperidine for the Discovery of a Potent NLRP3 Inflammasome Inhibitor”的文章,通过骨架杂交策略得到了一系列具有NLRP3抑制活性的丹参酮Ⅰ-哌啶杂化物,相较于丹参酮Ⅰ,这些化合物在活性、选择性和类药性具有显著改善。其中化合物5j、12a和12d对IL-1β的分泌有较强的抑制作用,在脓毒症小鼠模型中也具有较好的治疗效果。机制研究表明,这些化合物可以阻断ASC的寡聚化,抑制NLRP3炎症小体的激活,且化合物5j可与NLRP3蛋白直接结合,对NLRP3蛋白具有显著亲和力。本研究发现了一种全新结构的丹参酮Ⅰ衍生物,为NLRP3炎性体抑制剂的开发提供了新的思路。[/size] [size=14px] [/size] [size=14px] [/size] [size=14px]1、设计合成了36种Tan Ⅰ-哌啶杂化物前期研究发现Tan Ⅰ中的醌结构是其主要药效团,不宜进行结构修饰。因此研究团队从呋喃结构入手,通过支架杂交的策略,利用哌啶合成出了5个系列 36个Tan Ⅰ的衍生物。为了提升反应活性,在引入哌啶骨架前,研究团队将Tan Ⅰ中的醌并呋喃部分活化为富电子的苯并呋喃。[/size] [size=14px] [/size] [size=14px] [/size] [size=14px]2、体外生物学评价Tan Ⅰ-哌啶杂化物抗炎活性前期研究发现Tan Ⅰ具有抗炎活性,而NLRP3炎症小体作为炎症反应的核心,被证明与多种炎症性疾病相关。因此,作者选用小鼠腹膜巨噬细胞(PMs)开展了一系列体外生物学评价,首先通过MTT法发现36种Tan Ⅰ-哌啶杂化物在4 μΜ浓度无明显细胞毒性,随后发现与Tan-I相比,化合物5d、5j、10c、10f、10g、12a、12d在2 μΜ浓度下更能抑制IL-1β分泌,其中化合物12d与经典的NLRP3抑制剂MCC950活性相当。综合构效关系结果发现引入氢键受体或亲水基团可提升抑制活性(5j、12a、12d),于是作者选用化合物5j、12a、12d作为进一步的研究对象。[/size] [size=14px] [/size] [size=14px] [/size] [size=14px]3、化合物5j、12a和12d阻断NLRP3炎症小体激活,是广谱抑制剂NLRP3炎症小体通路包含准备和激活两个阶段,准备阶段pro-IL-1β和pro-caspase-1的表达升高,而激活阶段IL-1β和caspase-1分泌增加。作者发现化合物5j、12a、12d可抑制IL-1β和caspase-1的分泌,而对pro-IL-1β和pro-caspase-1的表达没有显著影响,表明它们通过阻断激活阶段而不是准备阶段来抑制NLRP3炎症小体活化。此外,5j、12a、12d也可以抑制尿酸钠晶体(MSU)、尼日利亚菌素(Nig)刺激的NLRP3炎症小体激活,表明5j、12a和12d是针对NLRP3炎症小体激活的广谱抑制剂。[/size] [size=14px] [/size] [size=14px] [/size] [size=14px]4、Tan Ⅰ-哌啶杂化物5j/12a/12d可抑制ASC寡聚化,5j可直接结合NLRP3蛋白ASC寡聚化可促进caspase-1的活化,是NLRP3炎症小体活化的标志之一。作者进一步研究化合物5j、12a、12d抑制NLRP3炎症小体的作用机制,通过免疫荧光实验发现在添加化合物5j、12a、12d和阳性药MCC950时,ASC寡聚化形成的斑点显著减少,表明它们均可抑制ASC寡聚化。接着利用表面等离子体共振分析(SPR)和细胞热位移测定(CETSA)实验证明化合物5j和NLRP3存在直接互作。[/size] [size=14px] [/size] [size=14px]5、Tan Ⅰ-哌啶杂化物在脓毒症小鼠模型的体内抗炎评价接着作者对Tan Ⅰ-哌啶杂化物5j、12a、12d进行了成药性评价,发现它们相较于Tan Ⅰ有极大的改善。进一步开展体内抗炎效果评价,发现在LPS诱导的炎症性脓毒症小鼠模型中,化合物5j、12a和12d预处理可以显著降低IL-1β的释放,显著改善肺组织病理损伤,如肺泡壁增厚明显减轻,粒细胞数量和炎症浸润显著减少。[/size] [size=14px] [/size] [size=14px] [/size] [size=14px]总结该研究通过骨架杂交策略得到了一系列具有NLRP3抑制活性的丹参酮Ⅰ-哌啶杂化物,与原型丹参酮Ⅰ相比,这些新的结构化合物在效力、选择性和类药性方面有显著改善,其中化合物5j、12a和12d对IL-1β的分泌具有高抑制活性。机制研究表明,这些化合物可以阻断ASC的寡聚化,抑制NLRP3炎症小体的激活,同时SPR和CETSA显示化合物5j可与NLRP3蛋白直接结合。体内研究表明它们对脓毒症小鼠模型具有较好的治疗效果,研究开发出了一种丹参酮I的简单结构修饰策略并提供了一类新的有效的NLRP3炎症小体抑制剂。[/size] [size=14px] [/size] [size=14px] [/size] [size=14px] [/size] [size=14px] [/size] [size=14px] [/size] [size=14px] [/size] [size=14px] [/size] [size=14px] [/size]
求助,谁有甲基汞乙基汞处理测试方法啊?
需要购买氯化甲基汞、氯化乙基汞的纯品标准,请提供相关的信息及联系方式。
求助二氟甲基-2,2,2-三氟乙基醚的标准红外谱图,谢谢上传。
各位[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]界的达人,求助各位一个问题: 有个氨基哌啶,成品盐酸盐,无紫外吸收,但也不想采用滴定的方法分析,那么,采用哪种比较适宜的GC固定性和条件分析这个氨基哌啶盐酸盐的含量呢?是否采用固定相为SE-54 或PEG20000的这两种不同极性的都可以呢?可检索到的关于哌啶或者哌啶盐酸盐的分析方法太少了啊。在此积极求助,忘各位指导迷津啊。
请问何处能提供SF-1150氰乙基甲基聚硅氧烷固定相?
ethyl methylketon是乙基甲基酮的意思么,也就是2-丁酮么
N-乙基-2-氨甲基吡咯烷如何检测水分
REACH SVHC 3-乙基-2-甲基-2-(3-甲基丁基)-1,3-恶唑烷 如何测试?可以用GCMS吗?前处理呢?
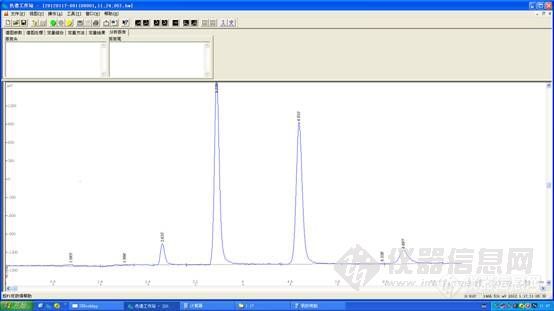
甲基,乙基和氯化汞形态分析 原子荧光与液相色谱联用,做混合标准溶液时,每天测的都不一样,差别极大,怀疑是溶液不稳定,或者污染,重新配制标准溶液,又有变化,十分不解?ps但是标准溶液都是参照方法用水直接溶解的,器皿都浸泡过。http://ng1.17img.cn/bbsfiles/images/2012/01/201201171633_346172_1619679_3.jpg
有谁做过1-丁基-3-甲基咪唑氯盐的分析?(1)色谱柱为 ODS 型C18色谱柱;流动相为水和甲醇,流动相比为水∶甲醇 = 1∶9,流速为1.0 mL/min;室温条件下检测,紫外检测波长为215 nm。(2) BDS型C18色谱柱;流动相为水(pH = 1.63)∶甲醇 = 85∶15,流速为1.0 mL/min;室温下检测,紫外检测波长为215 nm。我用第一种方法做过,在2.5分钟有一个很大的峰出来,但是接着就有一个很小的峰出来.两个峰分不开.
气相色谱分离甲基汞和乙基汞,用什么柱子好一点呢
求教用顶空做哌啶和DMF溶剂残留的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件设置,目前试过DB-624 ,HP-5柱子,db-624可以让两试剂出峰,但是哌啶出峰情况不稳定且很容易残留,峰面积时大时小,且混合样中甚至可能不出峰,DMF出峰情况很稳定,但是峰面积过小。试过提高顶空平衡温度,效果不佳。HP-5,DMF不出峰,哌啶出峰情况较DB-624好,峰面积稳定,拖尾不严重。
非药品类易制毒化学品分类和品种目录 第一类 1.1-苯基-2-丙酮 2.3,4-亚甲基二氧苯基-2-丙酮 3.胡椒醛 4.黄樟素 5.黄樟油 6.异黄樟素 7.N-乙酰邻氨基苯酸 8.邻氨基苯甲酸 第二类 1.苯乙酸 2.醋酸酐☆ 3.三氯甲烷☆ 4.乙醚☆ 5.哌啶☆ 第三类 1.甲苯☆ 2.丙酮☆ 3.甲基乙基酮☆ 4.高锰酸钾☆ 5.硫酸☆ 6.盐酸☆ 你们有不备案采购试剂的经历吗?如果有,你们采购到的试剂在外表/包装/质量上有什么区别吗?如果没有,那你们觉得备案麻繁琐吗?每次购买都需要备案吗?以上化学试剂那些是你们常买的?
应广大客户的需求,安谱公司推出欧盟REACH指令中的高关注度物质(SVHC)候选物质: 3-乙基-2-甲基-2-(3-甲基丁基)-恶唑烷对照品 3-Ethyl-2-methyl-2-(3-methylbutyl)-oxazolidine CDAA-380001-500mg CAS:143860-04-2 报价:1300.00 现货供应
春节好 求助氮氧自由基哌啶醇含量化验方法 谢谢!
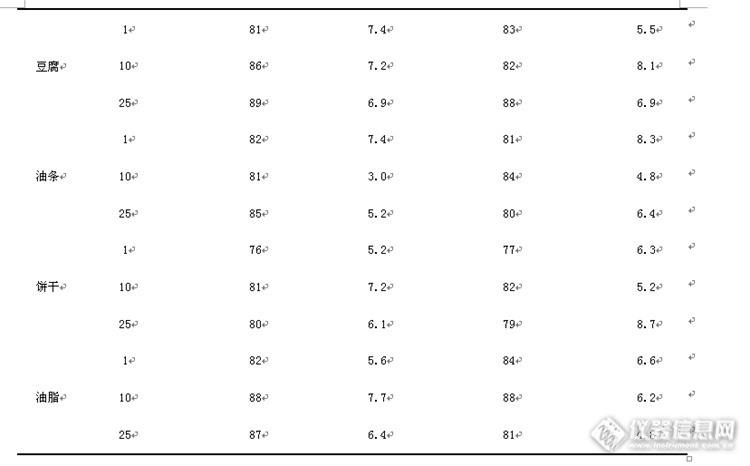
2014年底台湾惊爆二甲基黄食品安全事件,不法商贩将非食用色素二甲基黄添加到豆制品中着色牟利。二甲基黄及其同系物二乙基黄属于亲脂偶氮性染料,这种偶氮类物质多含有R-N=N-R键和其他芳香环或其衍生物的结构,被人体食用后易在肠道还原或分解为易致癌的芳香胺类,在此次台湾食品安全事件发生之前,偶氮染料如苏丹红、甲苯胺红、对位红等早已被禁止添加到食品中。除了豆制品外,粮食、油脂、油炸食品及饼干糕点都有可能为了追求色泽和降低成本而在生产过程中非法添加这两种偶氮染料。因此,如何在各类食品中有效、准确地检出这类非食用色素也成为当前的食品安全热点之一。本文尝试建立气相色谱质谱联用法对大米、豆腐、油条、饼干以及油脂中的二甲基黄和二乙基黄进行检测,在实现化合物有效分离的基础上,提高检测效率,为食品安全风险监测提供有效技术支撑。1 材料与方法1.1材料与试剂二甲基黄(≥98.5%,Dr.EhrenstorferGmbH)、二乙基黄(≥98.5%,Dr.EhrenstorferGmbH)、乙腈(色谱纯,Merck公司)、氯化钠(分析纯,国药集团化学试剂有限公司)、实验用水超纯水1.2 仪器与设备气相色谱-质谱联用仪:GCMS-QP2010, 日本岛津公司离心机:Centrifuge 5804R,德国Eppendorf公司超声波清洗器:KQ-500B型,昆山市超声仪器有限公司旋转蒸发仪:RE-2000A,上海亚荣生化仪器厂分析天平:BS224s,北京赛多利斯仪器系统有限公司涡混振荡仪:CM-1000,东京理化器械株式会社有油基质玻璃萃取管:上海安谱科学仪器有限公司1.3 方法1.3.1 色谱条件色谱柱:HP-5 MS,色谱条件:柱温: 40 ℃用于1分钟,30 ℃ /min升至180 ℃ (保持 3min),5 ℃ /min升至250 ℃,保持6min进样口:220 ℃分流方式:不分流1.3.2 质谱条件离子源为电子轰击离子(EI)源,电子轰击能量为70eV,离子源温度为230℃,四极杆温度为150℃,分别采用全扫描SCAN和选择离子SIM模式,溶剂延迟时间为5min。二甲基黄选择离子:77,105,120,225,二乙基黄选择离子:253,238,148,133。1.3.3 样品前处理大米、豆腐:称取4g,视含水量酌情加入少量去离子水后静置10min,加入2-3g NaCl后以10mL1%醋酸的乙腈提取并超声20min。振荡10min后以2500r/min离心5min。提取上清液,重复一次提取过程后收集上清液于45℃下浓缩至近干,并以1mL乙腈定容后上机。植物油:称取0.5g油脂,将其放入有油基质玻璃萃取管中,加入2mL 1%醋酸的乙腈涡混振荡2min后离心,将上清液上机。油条、饼干:称取4g样品,视含水量酌情加入少量去离子水后静置10min,加入少量NaCl后以10mL1%醋酸的乙腈提取并超声20min。振荡10min后以2500r/min离心5min。提取上清液,重复一次提取过程后收集上清液于45℃下浓缩至近干,以乙腈定容至2mL,转移至有油基质玻璃萃取管中,涡混2min后离心取上清液上机。2 结果与分析2.1 样品提取溶剂的选择在提取过程中,提取溶剂的选择对二甲基黄、二乙基黄的回收率有很大影响。根据二甲基黄的油溶性,分别选择丙酮、乙酸乙酯和乙腈为提取溶剂,通过比较回收率考察提取溶剂的合适程度。由下图可得,在样品含水的情况下,通常不使用与水混溶的提取溶剂,提取液中会含有大量水分,而影响浓缩效果。乙酸乙酯和丙酮的提取效率较好,但其易提取样品中的大分子物质,提取出的杂质较多,且丙酮不易与水分开,不易用盐析出其中的水分。乙酸乙酯在提取油性样品时,将一些非极性亲脂性干扰物质同时提取出来,杂质较多。乙腈不溶于油,能沉淀蛋白质,且提取的脂肪少,与样品混合匀浆后,虽然提取液中可能有水分,但较易用盐析出。而加入1%醋酸的乙腈其回收率高,且稳定。这可能是由于二甲基黄、二乙基黄是酸性化合物,低pH的环境可使得它不会发生离解和溶剂化作用,保持其稳定性。故本实验采用1%醋酸的乙腈作为提取溶剂。http://ng1.17img.cn/bbsfiles/images/2016/12/201612281034_01_2238288_3.png2.2 净化方法的选择二甲基黄和二乙基黄为阴离子酸性化合物,宜用反相SPE小柱对其进行萃取。当样品为大米及豆腐时,其基质简单,由于在前处理过程中每多增一步骤即有可能伴随目标物损失,故应在保证回收率的前提下尽量简化净化步骤。实验中加入少量去离子水后可活化分子状态,便于溶剂与微细试样反复接触萃取。加入NaCl促进两相分配,有效降低待测物对水相的亲和力。对于含油量较多的饼干、油条及油脂,净化的重点集中在油脂的去除。我们分别对低温冷冻法、PSA基质分散固相萃取以及氨基小柱固相萃取三种净化模式进行考察。结果表明,经过氨基小柱的净化效果略差,低温冷冻法和PSA基质分散固相萃取的净化效果相似,但耗时长,不利于风险监测时效性的提高。因此净化方式选择PSA基质分散固相萃取,在实验中我们选用上海安谱科学仪器有限公司的有油基质玻璃萃取管。这种萃取管最初用于邻苯二甲酸酯类的检测,除油效果较好。2.3 色谱分离条件选择根据文献,采用HP-5MS作为分离色谱柱。由于二甲基黄出峰较晚,优化仪器条件时将前面的升温速率提高,并放缓第二段升温速率。进行样品测定时,如满足以下条件则判断样品为阳性结果:1、色谱峰的保留时间与标准样品色谱峰的保留时间一致,且偏差在±2.5%之内,2、所选择的监测离子均出现,3、离子丰度比符合下表要求。http://ng1.17img.cn/bbsfiles/images/2016/12/201612281036_01_2238288_3.png在提取过程中,提取溶剂的选择对二甲基黄、二乙基黄的回收率有很大影响。根据二甲基黄的油溶性,分别选择丙酮、乙酸乙酯和乙腈为提取溶剂,通过比较回收率考察提取溶剂的合适程度。由下图可得,在样品含水的情况下,通常不使用与水混溶的提取溶剂,提取液中会含有大量水分,而影响浓缩效果。乙酸乙酯和丙酮的提取效率较好,但其易提取样品中的大分子物质,提取出的杂质较多,且丙酮不易与水分开,不易用盐析出其中的水分。乙酸乙酯在提取油性样品时,将一些非极性亲脂性干扰物质同时提取出来,杂质较多。乙腈不溶于油,能沉淀蛋白质,且提取的脂肪少,与样品混合匀浆后,虽然提取液中可能有水分,但较易用盐析出。而加入1%醋酸的乙腈其回收率高,且稳定。这可能是由于二甲基黄、二乙基黄是酸性化合物,低pH的环境可使得它不会发生离解和溶剂化作用,保持其稳定性。故本实验采用1%醋酸的乙腈作为提取溶剂。得到的标准物质及加标样品的TIC图及SIM图如下所示:http://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_01_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_02_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_03_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_04_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_05_2238288_3.png2.4 线性范围和检出限以2种色素的质量浓度为横坐标,色谱峰面积为纵坐标绘制标准工作曲线,二甲基黄的线性方程为y=1.257*105-5.579*104,二乙基黄的线性方程为y=1.444*105-5.651*104。结果表明在0.125-25ug/mL范围内,标准曲线线性关系良好,相关系数均大于0.99。以3倍信噪比计算检出限,二甲基黄的检出限为0.002mg/kg,二乙基黄的检出限为0.001mg/kg。2.5 回收率和精密度由于二甲基黄及二乙基黄浓度低于1ug/mL时即接近无色,实验中称样量为2-5g,故将加标浓度设为25mg/kg,10mg/kg和1mg/kg。加标方式:由于二甲基黄与二乙基黄是脂溶性物质,采用乙醇为油性模拟介质。将二甲基黄与二乙基黄分别溶于乙醇后,将已知浓度的溶液浸泡于大米和豆腐制品,超声并过夜。油脂类则直接称量一定质量的标准物质并超声溶于油脂。由下表可见,各组分测定结果的相对标准偏差在2.8%-10.1%,平均空白加标回收率为74%-93%,满足GB/T 27404-2008的相关要求。http://ng1.17img.cn/bbsfiles/images/2016/12/201612281044_01_2238288_3.pnghttp://ng
哌啶和乙醇

[align=center][img=,600,400]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091122509261_9069_932_3.jpg!w600x400.jpg[/img][/align]香兰素、甲基香兰素和乙基香兰素,均为广泛使用的可食用香料。有浓烈的奶香气息,在香荚兰的种子中可以找到,也可以人工合成,被广泛的运用到,蛋糕、奶粉、冰激凌等食品的制作中。今天我们就来做一下在奶粉和蛋白粉中香兰素、甲基香兰素和乙基香兰素的检测。[b]适用范围[/b]适用于奶粉、蛋白粉中香兰素、甲基香兰素和乙基香兰素的检测。[b]溶液的配置[/b]1)标准储备液:分别精确称取香兰素、甲基香兰素和乙基香兰素10mg,分别用乙腈溶解并定容到10mL,浓度为1000mg/L。2)20%甲醇:移取20mL的甲醇,用水定容至100mL。3)5%乙酸甲醇:移取5mL乙酸,用甲醇定容至100mL。4)50%乙腈:移取50mL的乙腈,用水定容至100mL。[b]提取步骤[/b]奶粉1) 移取1g样品,加入10mL的水,振荡,超声10min,离心10min(8000r/min),吸取上清液;2) 再加入10mL水,振荡,超声10min,离心10min(8000r/min),吸取上清液;3) 重复2)的过程,合并所有上清液,混匀,再次离心2min,待净化。蛋白粉移取0.5g样品,加入10mL的水振荡,8000rpm下离心,取澄清液,再重复2次,总计30mL水提取,待净化。[b]SPE净化步骤[/b]SPE柱:月旭WelchromP-SAX规格:150 mg/6mL。活化:5 mL 甲醇、5 mL 水,弃去;上样:待净化液15mL上样,控制流速,不宜过快,弃去;淋洗:6mL20%甲醇水淋洗,弃去。洗脱:15mL5%乙酸甲醇洗脱,收集于旋转蒸发瓶,并抽干小柱。复溶:洗脱液在45℃下减压蒸干,用50%乙腈定容至1mL。过0.22μm滤膜,上HPLC检测。[b]色谱条件[/b][color=#333333][/color]色谱柱:月旭UltimateXB-C18 4.6×250mm,5μm流动相:A-0.1%磷酸溶液,B-甲醇(A/B=70/30等度洗脱)流速:1.0mL/min柱温:30℃进样量:20μL检测波长:280nm[align=left][b]色谱图或者加标回收率结果[/b][/align][align=center][b][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091122549495_1545_932_3.jpg!w690x386.jpg[/img][/b][/align][align=center][color=#333333][img=,600,131]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091122584398_3527_932_3.png!w690x151.jpg[/img][/color][/align][align=center][color=#333333]图1.奶粉对照香兰素20mg/L、甲基香兰素5mg/L、乙基香兰素10mg/L图谱[/color][/align][color=#333333][/color][align=center][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123014375_3369_932_3.jpg!w690x386.jpg[/img][/color][/align][align=center][color=#333333]图2.奶粉粉样过柱图谱[/color][/align][align=center][color=#333333][/color][/align][align=center][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123043784_5835_932_3.jpg!w690x386.jpg[/img][/color][/align][align=center][color=#333333]图3.奶粉样加标香兰素20mg/L、甲基香兰素5mg/L、乙基香兰素10mg/L图谱[/color][/align][align=center][color=#333333][/color][/align][align=center][color=#333333][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123079138_6486_932_3.jpg!w690x386.jpg[/img][/color][/color][/align][align=center][color=#333333][color=#333333][img=,600,158]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123107145_4550_932_3.png!w690x182.jpg[/img][/color][/color][/align][align=center][color=#333333][color=#333333]图4.蛋白粉对照香兰素100mg/L、甲基香兰素5mg/L、乙基香兰素15mg/L图谱[/color][/color][/align][align=center][color=#333333][color=#333333][/color][/color][/align][align=center][color=#333333][color=#333333][color=#333333][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123138565_2990_932_3.jpg!w690x386.jpg[/img][/color][/color][/color][/align][align=center][color=#333333][color=#333333][color=#333333]图5.蛋白粉样过柱图谱[/color][/color][/color][/align][color=#333333][color=#333333][color=#333333][/color][/color][/color][align=center][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123171107_510_932_3.jpg!w690x386.jpg[/img][/align][align=center]图6.蛋白粉样加标香兰素400mg/kg、甲基香兰素20mg/kg、乙基香兰素60mg/kg图谱[/align][align=center][/align][align=center][img=,600,216]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123204485_8036_932_3.png!w561x202.jpg[/img][/align][color=#333333][/color][align=center]表1.奶粉香兰素、甲基香兰素和乙基香兰素过P-SAX小柱加标回收表[/align][align=center][/align][align=center][color=#333333][color=#333333][color=#333333][img=,600,162]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123255715_3868_932_3.png!w641x174.jpg[/img][/color][/color][/color][/align][align=center][color=#333333]表2.蛋白粉香兰素、甲基香兰素和乙基香兰素过P-SAX小柱加标回收表[/color][/align][color=#333333][color=#333333][color=#333333][b]相关产品信息[/b][/color][/color][/color][align=center][img=,600,407]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091123287959_7092_932_3.jpg!w690x469.jpg[/img][/align]







 我要推广仪器
我要推广仪器
 下载APP
下载APP