拉米夫定由英国葛兰素史克公司(GlaxoSmithKline)开发研制生产,1995年首次在加拿大、美国上市销售,1998年在中国上市,商品名贺普丁。目前,在我国市场上销售的拉米夫定及其复方制剂是由史克公司独家销售。拉米夫定及其片剂和口服液于1999年4月10日授权在中国获得药品行政保护,保护期将于2006年10月终止。拉米夫定全球专利将于2006年9月到期。拉米夫定是近年新上市的一种抗乙肝病毒新药,其英文名称为Lamivudine,又称3—TC。是一种核苷类抗病毒药,对人类免疫缺陷病毒和乙型肝炎病毒均具有显著的抑制活性,是治疗艾滋病和乙型肝炎的具有显著疗效的药物。拉米夫定是第一个经美国FDA以及中国食品药品监督管理局批准的口服抗乙肝病毒药物。由于拉米夫定问世时间早,很长一段时间都是临床药物研究的“宠儿”,又由于其临床相对比较安全,应用较为广泛。现阶段,拉米夫定是乙肝病毒感染的一线治疗药物。拉米夫定的缺点:拉米夫定易出现耐药现象。但并未影响该药的销售额。据中国医药商业协会最新报道,2003年全国 28家主要医药批发公司畅销药品中,拉米夫定排名第13位,销售额为3.06亿元,在46种抗感染药品中占4.33%。2002年贺普丁世界性销售额为2.95亿英镑,2003 年为2.93亿英镑,折合4.8亿美元。2003年,贺普丁在我国16城市样本医院销售额排名41位,用药金额8011万元,其中进口药品占60.62%,是英国葛兰素和WELLCOME(UK)两家公司,该药在全国市场的销售额约为8亿多元。2004年为7311.43万元,仍占全身用抗病毒药物的38%,而在全国医院销售额已达到6亿~8亿元的市场规模。2005年上半年样本医院购入药物是苏州葛兰素史克、英国葛兰素史克和WELLCOME(UK)公司的药物,各厂商分别占据了51.03%、47.92%和1.05%的份额。迄今为止,全球每年约有80万人使用贺普丁,拉米夫定目前在中国的终端销售额达到10亿元人民币左右,在中国药品市场单品销售排行榜上位列第二。 拉米夫定原料药及其制剂目前均为进口药品。目前原料药及其制剂,国内尚未有生产厂家上市销售,也未有公司进行国内新药申报。另外英国GlaxoSmithKline公司在中国分公司(葛兰素史克制药(苏州)有限公司)也在中国已经获得生产批件,批准文号:国药准字H20030581。目前国内有,拉米夫定原料药以及拉米夫定片(两种规格)、拉米夫定口服液(两种规格)两种进口剂型上市销售。
阿德福韦酯是由美国Gilead Science公司开发的新型核苷类抗乙型病毒性肝炎药物,已在国外进行了Ⅱ、Ⅲ期临床试验。国外的临床研究资料表明,阿德福韦能有效地抑制HBV DNA的复制,使HBV DNA滴度迅速降低,而且在出现拉米夫定耐药的患者中阿德福韦能继续有效地抑制变异株。我国药品监督管理局于2000年12月批准该药在中国进行临床试验,目前,Ⅰ期临床试验已结束,Ⅱ期临床试验也已在2002年12月正式启动。 一、作用机制 阿德福韦酯是腺嘌呤磷酸酯化合物阿德福韦的前药,其分子式为C20H32N5O8P,分子量为501.48。口服后,在体内转化为阿德福韦发挥抗病毒作用。阿德福韦是单磷酸腺苷的核苷酸类似物,在体内通过细胞激酶作用被磷酸化为具有活性作用的二磷酸阿德福韦,二磷酸阿德福韦抑制HBV DNA多聚酶或逆转录酶作用机制包括:(1)竞争脱氧腺苷三磷酸底物;(2)终止病毒DNA链延长。二磷酸阿德福韦对HBV DNA多聚酶的抑制常数为0.1mmol/L;对人类DNA多聚酶α和γ的抑制作用较弱,其抑制常数分别为1.18mmol/L和0.97mmol/L,因此,治疗剂量对正常细胞没有毒性。 二、药效和毒理 在体外实验中,阿德福韦抑制HBV转染人肝细胞瘤细胞株HepG2和HB611细胞病毒复制的半数抑制浓度(IC50)分别为0.2~2.5mmol/L和0.2~1.2mmol/L。二磷酸阿德福韦在细胞内的T1/2为30h,故作用较持久,可以每天给药一次。 拉米夫定耐药株涉及HBV DNA聚合酶M552V、M552I、L528M、L552M/M552V位点的突变。在体外实验中发现这些突变体对阿德福韦仍敏感,与野生株比较,它们的抑制常数增加了不到2.2倍,而拉米夫定对变异株的抑制常数则增加了8~25倍。这些资料表明,阿德福韦可以治疗对拉米夫定耐药的HBV,而且与拉米夫定联合治疗可以有效控制对拉米夫定的耐药。同时,体外实验也发现阿德福韦对泛昔洛韦耐药株也较敏感。 在体内实验中,发现阿德福韦能有效地抑制鸭乙型肝炎病毒和土拨鼠肝炎病毒(WHV)的复制。给予慢性感染WHV的成年土拨鼠每日口服5mg/kg、15mg/kg的阿德福韦或安慰剂治疗12周,治疗后5mg/kg剂量组WHV DNA水平减少了260倍,而15mg/kg剂量组则下降了1000倍以上。 四、国外临床研究进展 阿德福韦已在美国、欧洲、澳大利亚及东南亚进行了Ⅱ、Ⅲ期临床试验。临床试验涉及HBeAg阳性和考虑有前C区变异的HBeAg阴性的慢性乙型肝炎患者、对拉米夫定耐药的代偿性肝病患者、合并人类免疫缺陷病毒感染的拉米夫定耐药患者、肝移植前或移植后对拉米夫定耐药的失代偿性肝病患者。 早期进行的针对HBeAg阳性、ALT异常或正常的二项双盲、安慰剂对照的Ⅱ期临床试验,疗程为12周,并随访24周。ALT异常临床研究的患者,接受剂量为5mg/d、30mg/d和60mg/d。治疗12周后,5mg剂量组血清HBV DNA较基线下降1 Log10,30mg和60mg剂量组血清HBV DNA下降3~4 Log10,而安慰剂组无显著变化;36周后,30mg和60mg剂量组HBeAg转阴率为27%,HBeAg血清转化率为20%,血清转化率增高与基线时ALT水平呈正相关。ALT正常的临床研究患者,接受剂量为30mg/d。治疗12周后,30mg剂量组血清HBV DNA较基线下降3Log10,而安慰剂组无显著变化。所有接受治疗的患者在治疗12周后进行基因检测,没有发现与阿德福韦耐药有关的变异产生。在这二项研究中,30mg和60mg剂量组均出现部分患者的肾功能损害,表现为尿素氮和肌酐的升高,出现肾功能损害的比例与剂量呈正相关,故在以后的延续试验中以10mg剂量组而代替60mg剂量组。 在一项随机、双盲、安慰剂对照的临床试验中,共有515例HBeAg阳性的患者进入研究。在前48周,患者被随机分入阿德福韦30mg组(173例)、阿德福韦10mg组(172例)或安慰剂组(170例)。48周后,30mg组接受安慰剂治疗至96周,安慰剂组接受阿德福韦10mg治疗至96周,10mg组则再次随机按1:1接受安慰剂或继续阿德福韦10mg治疗至96周。所有患者在第一次随机前6月内接受第一次肝活检,在治疗48周、96周后接受第二、三次肝活检。所有患者治疗96周后随访24周。治疗48周后,10mg组和30mg组组织学改善率(组织学改善定义为Knodell坏死炎症计分下降32分,且Knodell肝纤维化计分无恶化)分别为53%和59%,显著高于安慰剂组25%;10mg组和30mg组治疗后血清HBV DNA较基线时下降3.52 Log10和4.76 Log10,安慰剂组为0.55 Log10;10mg组HBeAg阴转率为24%,HBeAg血清转化率为12%,显著高于安慰剂组的6%和11%;10mg组ALT复常率为48%,安慰剂组则为16%。研究中,发现基线ALT水平与肝组织学改善和HBeAg血清转化呈正相关。另一项随机、双盲、安慰剂对照的临床试验,共有185例考虑有前C区变异的HBeAg阴性的患者按2:1比例进入阿德福韦10mg组或安慰剂组。治疗48周后,10mg组组织学改善率为64%,显著高于安慰剂组33%;10mg组治疗后血清HBV DNA较基线时下降3.91 Log10,51%患者HBV DNA转阴,安慰剂组HBV DNA较基线时下降1.35 Log10,没有患者HBV DNA转阴;10mg组ALT复常率为72%,著高于安慰剂组29%。目前本项研究仍在进行中。 五、耐药和病毒变异 阿德福韦较少产生耐药的分子学基础包括:(1)阿德福韦与自然底物dATP在结构上非常相像;(2)阿德福韦具有灵活的开链连接;(3)具有磷酸键。 629例患者在治疗48周后接受了病毒变异的检测,结果未发现产生阿德福韦耐药的病毒变异。2003年美国肝病年会上,Gilead公司报道,238例患者在治疗96周时有4例发现N236T位点的变异,发生率为1.7%,并证实N236T变异与阿德福韦耐药有关。另一可能与阿德福韦耐药有关的A181V位点突变,96周时的发生率为0.8%。另外一项研究是早期进行的针对HBeAg阳性、ALT异常或正常的二项双盲、安慰剂对照研究的延续。患者在治疗中没有出现血清转化,也没有出现与治疗相关的毒性反应,患者自愿继续接受治疗。剂量开始为30mg/d,后改为10mg/d。在长达136周的观察中,阿德福韦对野生株和前C区变异的慢性乙型肝炎具有持续的抗病毒作用,而且没有发现与阿德福韦耐药相关的病毒变异。 七、国内的研究状况 我国食物药品监督管理局于2000年12月批准该药在中国进行Ⅰ期临床试验。2001年6月~9月进行Ⅰ期临床试验,Ⅰ期临床试验包括3个研究方案:(1)在健康中国男性志愿者中,对单次口服阿德福韦片剂的安全性和耐受性进行评估的一项Ⅰ期、单中心、随机、双盲、安慰剂对照的研究;(2)在健康中国男性志愿者中,对阿德福韦片剂的药代动力学进行评估的一项Ⅰ期、单中心、开放、拉丁方设计的研究;(3)在健康中国志愿者中,就连续6d,1次/d,口服阿德福韦片剂的安全性、耐受性和药代动力学进行评估的一项Ⅰ期、单中心、随机、双盲、安慰剂对照的研究。I期研究结果显示在健康中国志愿者中口服阿德福韦片剂的安全性、耐受性良好;10mg剂量下,未观察到肾功能损害;药代动力学参数与国外研究结果相似。2002年10月国家药品监督管理局批准该药在中国进行Ⅱ期临床试验。Ⅱ期临床试验在中国的总病例数为480例,均为HBeAg阳性、HBV DNA阳性、ALT增高的患者。全国有12个中心参与。目前,Ⅱ期临床试验已在2002年12月正式启动,2003年2月底已完成最后一例患者入组。
我需要拉米夫定的标准红外谱图!自己做了一张,但是和客户的那一张对不起来,所以恳请好心人帮忙了.如果有此标准谱图的请发到我的邮箱:wuai-111@163.com先谢谢了!
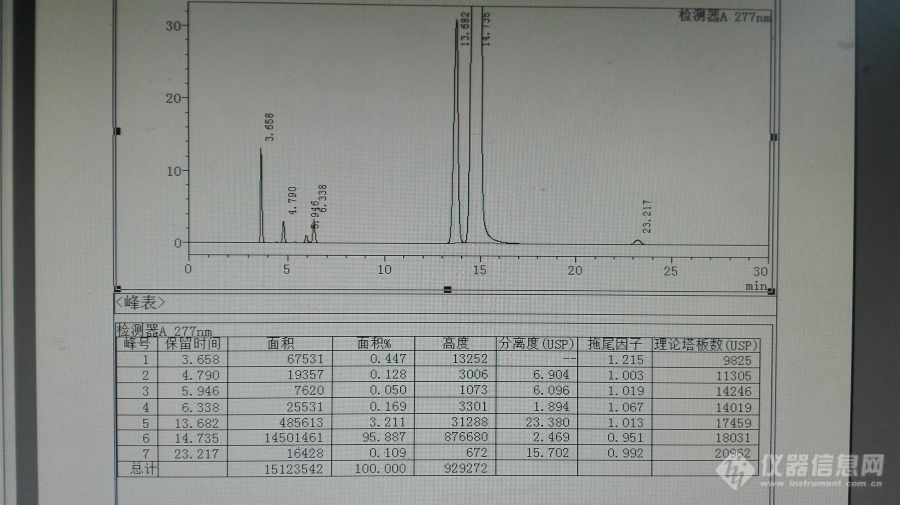
来自中国的自信,月旭Welchrom C18测定拉米夫定有关物质[b]除非经大量试验比较市面主流厂家的色谱柱均不能达到要求,最好不要把色谱柱的品牌定下来。[/b]拉米夫定(Lamivudine)对乙型肝炎病毒和HIV有明显的抑制作用。口服吸收后,拉米夫定可在HIV感染细胞和正常细胞内代谢生成拉米夫定三磷酸盐,它是拉米夫定的活性形式。后者通过竞争抑制作用,终止DNA链的延长,从而抑制HIV和HBV的反转录酶和HBV聚合酶,阻止HIV和HBV的DNA合成和病毒复制。体外实验中与齐多夫定联合,对HIV病毒有协同作用。下面为中国药典2015年版收载的质量标准,其中提到的色谱柱,我们咨询了价格比较贵,我们就用手上的月旭色谱柱进行了探索研究。参照了EP8.0质量标准,进行了系统适用性试验,其中EP8.0质量标准有关物质项下为:Related substances. Liquid chromatography (2.2.29).Test solution. Dissolve 50.0 mg of the substance to beexamined in the mobile phase and dilute to 100.0 mL withthe mobile phase.Reference solution (a). Dilute 1.0 mL of the test solution to100.0 mL with the mobile phase. Dilute 1.0 mL of this solutionto 10.0 mL with the mobile phase.Reference solution (b). Dissolve 5 mg of salicylic acid R in themobile phase and dilute to 100.0 mL with the mobile phase.Dilute 1.0 mL of the solution to 100.0 mL with the mobilephase.Reference solution (c). Dissolve 50.0 mg of lamivudine CRSin the mobile phase and dilute to 100.0 mL with the mobilephase.Reference solution (d). Dissolve 5 mg of cytosine R and 5 mg ofuracil R in the mobile phase and dilute to 100.0 mL with themobile phase. Dilute 2.0 mL of the solution to 10.0 mL withthe mobile phase.Reference solution (e). Dissolve 5 mg of lamivudine for systemsuitability 1 CRS (containing impurities A and B) in 2 mL ofthe mobile phase. Add 1.0 mL of reference solution (d) anddilute to 10.0 mL with the mobile phase.Column:- size: l = 0.25 m, Ø = 4.6 mm - stationary phase: base-deactivated octadecylsilyl silica gel forchromatography R (5 μm) - temperature: 35 °C.Mobile phase: mix 5 volumes of methanol R and 95 volumes ofa 1.9 g/L solution of ammonium acetate R, previously adjustedto pH 3.8 with glacial acetic acid R.Flow rate: 1.0 mL/min.Detection: spectrophotometer at 277 nm.Injection: 10 μL.Run time: 3 times the retention time of lamivudine.Identification of impurities: use the chromatograms obtainedwith reference solutions (b) and (e) to identify the peaks dueto impurities A, B, E, F and C.[b]Relative retention with reference to lamivudine (retentiontime = about 9 min): impurity E = about 0.28 impurity F = about 0.32 impurity A = about 0.36 impurity B = about 0.91 impurity J = about 1.45 impurity C = about 2.32.[/b]System suitability: reference solution (e):- resolution: minimum 1.5 between the peaks due toimpurities F and A minimum 1.5 between the peaks dueto impurity B and lamivudine.Limits:- correction factors: for the calculation of content,multiply the peak areas of the following impurities bythe corresponding correction factor: impurity E = 0.6 impurity F = 2.2 impurity J = 2.2 - impurity A: not more than 3 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.3 per cent) - impurity B: not more than twice the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.2 per cent) - impurity C: not more than the area of the principal peakin the chromatogram obtained with reference solution (b)(0.1 per cent) any other impurity: for each impurity, not more than thearea of the principal peak in the chromatogram obtainedwith reference solution (a) (0.1 per cent) - total: not more than 6 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.6 per cent) - disregard limit: 0.5 times the area of the principal peak inthe chromatogram obtained with reference solution (a)(0.05 per cent).中文名拉米夫定外文名lamivudineCAS号134678-17-4分子式C8H11N3O3S[img=,690,400]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302020_01_1621890_3.png!w690x400.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302026_01_1621890_3.png!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302027_01_1621890_3.png!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302028_01_1621890_3.png!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302029_01_1621890_3.png!w690x388.jpg[/img]以上均为参照中国药典2015年收载的拉米夫定片质量的截图,然后进行系统适用性试验,进行流动相微调,其典型色谱图见下图:[img=,690,353]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302137_01_1621890_3.png!w690x353.jpg[/img]初步统计色谱峰保留时间,其他色谱图参数很好,理论板数都超级好,分离度和拖尾因子也超级好! [table=167][tr][td]相对保留时间(RRT)[/td][td]保留时间(min)[/td][/tr][tr][td]0.25 [/td][td]3.658[/td][/tr][tr][td]0.33 [/td][td]4.79[/td][/tr][tr][td]0.40 [/td][td]5.946[/td][/tr][tr][td]0.43 [/td][td]6.338[/td][/tr][tr][td]0.93 [/td][td]13.682[/td][/tr][tr][td]1.00 [/td][td]14.735[/td][/tr][tr][td]1.58 [/td][td]23.217[/td][/tr][/table]其中水杨酸的色谱峰如下:[img=,690,338]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302144_01_1621890_3.png!w690x338.jpg[/img]另外最好控制柱温,最好是液相自动控温装置,不要外接质量差的柱温箱,因为在升温的过程有±5℃的差异,会引起水杨酸色谱峰保留时间和峰型变化较大,使其用相对保留时间定位出现偏差,建议用水杨酸对照品同时定位,因为其价格便宜易得,其典型色谱图如下:[img=,690,320]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302159_01_1621890_3.png!w690x320.jpg[/img]拉米夫定对照品定位溶液主峰典型色谱图如下:[img=,690,405]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302150_01_1621890_3.png!w690x405.jpg[/img][img=,690,409]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302211_01_1621890_3.png!w690x409.jpg[/img]以上拉米夫定对照品出峰保留时间基本上变化不大,但是浓度较大是有较大差异,15分钟在14.4分钟变化。其变化情况见下面系统色谱图。但是其相对于水杨酸的变化幅度较小。系统进针另外图谱:[img=,690,375]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302207_01_1621890_3.png!w690x375.jpg[/img][img=,690,415]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302209_01_1621890_3.png!w690x415.jpg[/img]总结:我们没有用到中国药典的色谱柱,但是从色谱图及其系统适用性试验结果,其系统各杂质均能有效分离,和EP8.0提到的主峰保留时间差距很大,约6分钟差异,估计是色谱柱填料性能差异引起,从研究角度来讲,[b]中国产的色谱柱完全适用于本品的有关物质检测和含量测定,为我们后续的研究增强了自信心。作为权威的中国药典,不知道什么原因把色谱柱的品牌给定下来了,个人愚见,除非经大量试验比较市面主流厂家的色谱柱均不能达到要求,最好不要把色谱柱的品牌定下来。作为国产优良色谱柱的生产商月旭公司不得不由衷给个赞![/b]

LC-MSMS做拉米夫定标准品,不知为啥一直有个明显肩峰。图谱在附件里http://ng1.17img.cn/bbsfiles/images/2013/09/201309051141_462516_2167114_3.bmp
现在有一课题,是做氨基酸原料药的杂质研究,这到底是指氨基酸类杂质,还是所有可能出现的杂质,有点蒙圈,感觉好难
本人在做氨基酸分析,用的是黄酒中氨基酸的测定方法行业标准编制说明 标准,十八种氨基酸混标进样,由于仪器限制,使用的是二元梯度,标准是三元,两者的区别只是峰的分离度。但是实验中总是有杂质峰,而且重复性很好,保留时间、峰面积是一致的,怀疑是衍生副产物。单标进样仍然有,而且几乎是一个单标对应一个杂质峰。并不是所有杂质峰都出现,说明不是样品、流动相的问题。认为问题就出在衍生过程中,有做过的是否也由这些杂质峰?
氨基柱通常都是用不低于50%的有机相冲洗柱子,这样能否保证杂质洗脱的干净呢?
我HPLC测糖蜜和玉米浆(含杂质较多,蛋白、多种氨基酸等)中生物素含量,标品中生物素在7.7分钟出峰,在两个样品中7.7分时间左右有风出现,但峰面积很大,经计算不合实际,初步判断不是生物素的峰,可能是被杂峰覆盖,请问该怎么处理???另外可能由于糖蜜和生物素中生物素含量低,没有峰出现,但经手动积分事件仍没有峰出现,因此此种可能被排除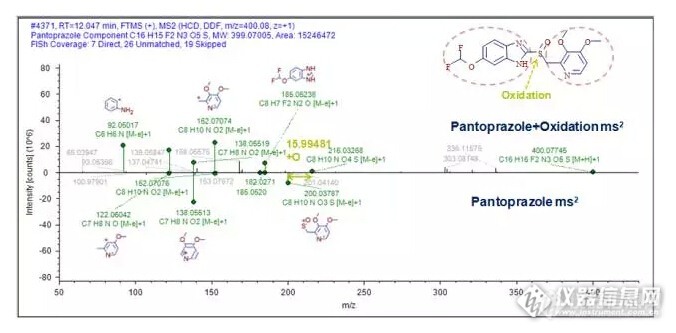
药物杂质是药物活性成分(原料药)或药物制剂中不希望存在的化学成分,会对用药的安全性和有效性带来隐患,因此杂质的检测是保证药物质量至关重要的部分,FDA、EMEA、PMDA、CFDA等各国药品监管部门制定了相应的指导原则对其进行严格管控。http://ng1.17img.cn/bbsfiles/images/2015/12/201512141737_577892_3005330_3.jpg 独有的四极杆静电场轨道阱Q Exactive™ Focus高分辨液质联用技术,凭其高灵敏度、高专属性和高准确性的分析能力,可对样品中药物杂质进行全面的信息采集。结合新一代的智能小分子化合物鉴定软件Compound Discoverer™,以高度灵活的自定义方式制定分析工作流程,对数据中的目标和非目标杂质进行提取、比对及鉴定,工作流程如下:http://ng1.17img.cn/bbsfiles/images/2015/12/201512141737_577893_3005330_3.jpg 通过软件对样品数据的分析和提取,在Compound Discoverer中可以直观、便捷的查看和筛选预期和未知的杂质分析结果,从结果界面中可获得不同条件下样品杂质的变化情况,获得所有杂质保留时间、一级质谱、同位素和二级质谱等丰富信息:http://ng1.17img.cn/bbsfiles/images/2015/12/201512141738_577894_3005330_3.jpg 在获得母药和杂质的一级和二级质谱信息后,软件将调用碎裂数据库(Fragmentation Library)快速的对泮托拉唑的碎片结构进行归属,该数据库几乎涵盖了所有已发表的文献,保证了碎片解析的准确性。在此研究结果之上,通过软件对杂质与母药二级质谱信息之间的比对,可进一步对杂质变化位点进行推测。在本例中,通过152、185等共有碎片和200、216等特征差异碎片的比对,推测出该杂质为泮托拉唑砜:http://ng1.17img.cn/bbsfiles/images/2015/12/201512141738_577895_3005330_3.jpg 基于新一代四极杆-静电场轨道阱质谱Q Exactive Focus和新一代小分子化合物分析软件Compound Discoverer,建立了药物杂质鉴定的新流程。无论是优质数据的有效获取,还是获取后对已知和未知杂质的分析鉴定,该工作流程都可以完美的实现。在本例中,共鉴定到泮托拉唑杂质15个,其中可能的降解杂质9个,可能的工艺杂质6个,为药物杂质的质量控制、安全性评估提供了富有价值的信息。(分享)
本人在做氨基酸分析,用的是黄酒中氨基酸的测定方法行业标准编制说明 标准,十八种氨基酸混标进样,由于仪器限制,使用的是二元梯度,标准是三元,两者的区别只是峰的分离度。但是实验中总是有杂质峰,而且重复性很好,保留时间、峰面积是一致的,怀疑是衍生副产物。单标进样仍然有,而且几乎是一个单标对应一个杂质峰。并不是所有杂质峰都出现,说明不是样品、流动相的问题。认为问题就出在衍生过程中,有做过的是否也由这些杂质峰?应该如何处理这些杂质峰?
同一瓶样品,重复称样、定容、手动进样8次。C18柱流动相:按药典缓冲盐:乙腈(80:20),主峰前有3个杂质峰,主峰后有一个,只是第一个杂质峰面积不稳定,上下波动。第一个杂质峰第一针峰面积156;第一个杂质峰第一针峰面积52第一个杂质峰第一针峰面积49第一个杂质峰第一针峰面积40第一个杂质峰第一针峰面积57第一个杂质峰第一针峰面积32第一个杂质峰第一针峰面积50第一个杂质峰第一针峰面积49又对另一个批次样品进样2针(重复称样)第一个杂质峰第一针峰面积 159第一个杂质峰第一针峰面积 83老师们分析下,到底是系统问题还是样品中相关杂质不稳定?又该如何调整?跪求答复!进样前基线和空白对照都很好!不同样品间都走空白,空白此杂质的积分面积只有1个单位!可以忽略1
我用的是日立D-2000高效液相色谱仪器 新的 新的waters 氨基柱子 用错了流动相是 乙二胺四乙酸而那钙盐 ,用的时间很短,后来用水冲洗了很久,又用了乙腈。每次出现的是杂质峰,一大一小。太强了信号(700左右),主峰都看不到啊,杂质峰出峰时间固定,面积固定。。现在怎么办啊。。进什么标样出来的杂质峰都一样啊。。一样看不到主峰在哪儿。进样针进行了多次的清洗。进了多次的乙腈都是倒峰。
为了测定邻氨基苯酚中微量杂质苯胺,现有下列固定相:硅胶,ODS键合相,流动相有。水-甲醇,异丙醚-己烷,应选用哪种固定相,流动相?为什么?
我现在用2010CP标准检测对氨基水杨酸钠液相,怎么间氨基酚和单一杂质都分不出来,请问2010CP标准的流动相配比是不是有问题?
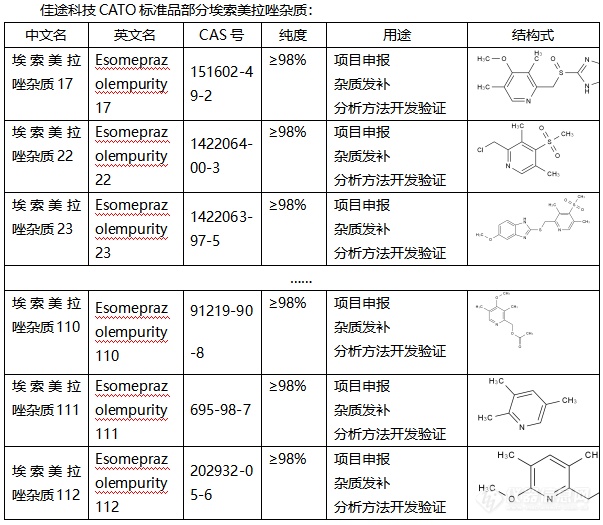
◇关于埃索美拉唑杂质 埃索美拉唑杂质是一种质子泵抑制剂,它不仅是[font=UICTFontTextStyleBody]治疗胃食管反流性杂质,还可以防止胃酸形成,[/font]它的原理主要是通过抑制胃壁细胞中[font=.pingfang sc]的[/font]H+/K+-ATP酶来达到减少胃酸分泌。埃索美拉唑杂质是一种高效且广泛应用于胃酸相关疾病治疗的质子泵抑制剂,通过抑制质子泵的活性,它不仅可以减少胃酸的分泌,还可以帮助溃疡的愈合。其作用机理是通过与胃腺细胞内的质子泵结合,形成稳定的复合物来发挥作用。[font=UICTFontTextStyleBody] [/font][font=UICTFontTextStyleBody]CATO[/font]标准品提供的[font=宋体]埃索美拉唑杂质[/font][font=宋体],在治疗肠胃道疾病中发挥着重要的作用,并且有针对性的抗菌作用。[img=,603,525]https://ng1.17img.cn/bbsfiles/images/2024/02/202402040930531289_1324_6381607_3.png!w603x525.jpg[/img] [/font]
下面的结构式是富马酸卢帕他定的一个氧化杂质,我想请问下这是哪个化合物,N旁边的O是怎么结合的?[img]https://ng1.17img.cn/bbsfiles/images/2021/10/202110201856032930_4323_3860760_3.png[/img]
富马酸沃诺拉赞在当前常被认为钾离子竞争性的酸阻滞剂(P-CAB),属于一种当前新推出的可逆性质子泵抑制剂。富马酸沃诺拉赞片是武田制药公司研制,于2014年首次在日本获批上市,主要用于治疗幽门螺杆菌感染、胃食管反流、胃溃疡、消化性溃疡等胃酸相关性疾病。2018年7月在韩国上市,2019年12月在中国获批上市。由于富马酸沃诺拉赞药物原型发挥作用,起效迅速,且半衰期长,抑酸作用显著而受到广泛关注。CATO标准品提供的富马酸沃诺拉赞杂质标准品,是用于药物分析和质量控制的化学物质。

依托咪酯是一种麻醉药物,用于全身麻醉。在生产过程中,可能会产生一些杂质。这些依托咪酯依托咪酯的杂质可能包含化学反应的副产物、落在设备上的残留物、原料中的不纯物,等等。杂质过多可能会影响药物的质量、效力和安全性。比如,某些杂质可能会引导药物的疗效降低,或者引发不良反应。因此,检测和控制杂质是制药工艺中的一个重要环节。通过严格的质控程序,可以将杂质的量控制在安全的范围内,以保证药物的效力和安全性。CATO标准品对依托咪酯的杂质进行研究和检测,也可以帮助我们理解并改进制药过程,找出可能产生杂质的环节,进行优化,从而提高药品的质量和效力[img=,612,514]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052108155537_3951_6381668_3.png!w612x514.jpg[/img]
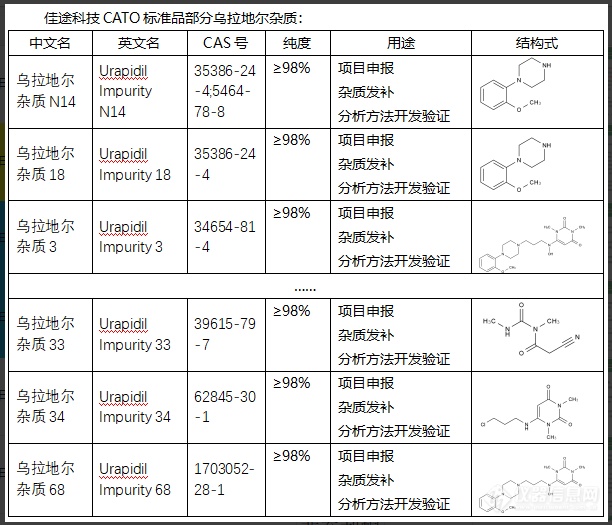
乌拉地尔(Uradil)是RNA分子中的一种核苷酸,通常聚合在RNA链上对编码蛋白质起重要作用。然而,乌拉地尔杂质通常被认为是DNA序列中的错误或错误的插入。乌拉地尔杂质的存在可能会导致DNA复制和转录的错误,从而导致基因表达的改变或突变,进而可能导致细胞功能异常,诱发一些健康问题,比如癌症等。由于这个原因,CATO标准品生物体内有专门的机制,例如尿苷DNA糖苷酶,可以检测并修复DNA中的乌拉地尔杂质,以维持DNA的稳定性和遗传信息的准确性。[img=,612,525]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041442469549_3655_6381668_3.png!w612x525.jpg[/img]
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]
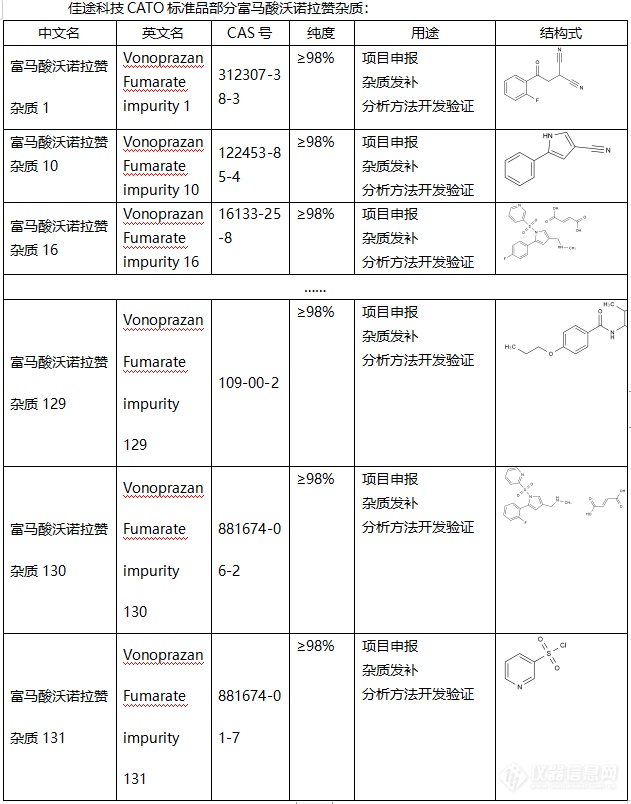
◇关于[font=UICTFontTextStyleBody]富马酸沃诺拉赞杂质[/font][font=UICTFontTextStyleBody][/font] 富马酸沃诺拉赞是一种抑酸的药物,又称为钾离子竞争性酸阻滞剂,通过[font=.pingfang sc]竞争性的阻断[/font]H+,K+-ATP酶(质子泵)K+结合位点,抑制了K+对H+,K+-ATP酶(质子泵)的结合作用,从而达到抑制了胃酸分泌的效果,除此之外还可以抑制胃肠道上部黏膜损伤的形成,在临床上可以治疗反流性食管炎。富马酸沃诺拉赞与普通抑制胃酸的药物,例如剂奥美拉唑、兰索拉唑等相比较,本品因为无需肠溶包衣,所以奇效更快,效果时间也更长。 [font=UICTFontTextStyleBody]CATO[/font]标准品提供的[font=UICTFontTextStyleBody]富马酸沃诺拉赞杂质[/font],可以治疗胃溃疡、十二指肠溃疡等疾病。[font=UICTFontTextStyleBody][font=.pingfang sc] [/font][/font][img=,631,804]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021602111695_2371_6381607_3.png!w631x804.jpg[/img][font=UICTFontTextStyleBody] [/font]
请问重氮乙腈的分析方法。(其中所含杂质为:氨基乙腈 ,亚硝酸钠,亚硝酸)
普拉克索杂质A,B,C,D,E欧洲药典标准。进口注册标准中代码【BI-II751XX】 【BI-II786BS】 【BI-II820BS】BI-II 546 CL】常用杂质对照品
[align=center]奥美拉唑成品中杂质的质量检测[/align][align=center][b]摘要:目的:[/b]在对奥美拉唑原料药中引入的基因毒性杂质,即4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺设定质量检测方法,并进行方法验证。同时,对合成工艺中引入的残留溶剂进行质量检测,确保奥美拉唑成品的质量安全。[b]方法:[/b]在对4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺进行质量检测中,采用液相色谱的方法,并对其进行限度验证;而对残留溶剂采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],利用内标法进行质量检测。[b]结果:[/b]本研究对两种基因毒性杂质及残留溶剂的检测是可行有效的,有利于对奥美拉唑原料药的质量监控,同时为后续对奥美拉唑质量标准的制定提供理论依据。[b]关键词:[/b]奥美拉唑;质量标准;毒性杂质;残留溶剂[/align][b]Abstract Objective[/b]: Themass detection is set in the introduction of genotoxic impurities into theomeprazole APIproducts, namely 4-methoxy-2-nitroaniline and4-methoxy-o-phenylenediamine, and the method was verified.At the same time, the quality of theresidual solvent introduced in the synthesis process is checked to ensure thequality and safety of the omeprazole.[b]Methods:[/b]In the mass detection of4-methoxy-2-nitroaniline and 4-methoxy-o-phenylenediamine, the method of liquidchromatography is used, and the limit is verified the residual solvent aretested by the gas chromatogram and internal standard method for quality.[b]Results:[/b]This study is feasible and effective for the detection of twogenotoxic impurities and residual solvents, which is the benefit of qualitymonitoring of omeprazole APIproducts, and provides a theoretical basis forthe subsequent development of omeprazole quality standards.[b]Keywords:[/b] Omeprazole Quality standard Genotoxicimpurities Residual solvents随着人们平时工作、学习等压力的不断增加,导致消化类疾病患病率不断上升,而在中国,发病率已达到20%左右[sup][/sup]。用于治疗消化类疾病的药物也逐步成为生活中的常用药,其发展市场也在不断扩大。在消化系统溃疡类疾病的临床治疗中,质子泵抑制剂类药物因其具有良好的治疗效果,市场销售份额高达58%[sup][/sup]。而奥美拉唑是质子泵抑制剂类的代表药物,通过抑制胃酸分泌,用于治疗胃溃疡、十二指肠溃疡等疾病。但长期服用奥美拉唑存在着潜在风险,可能会引起心脏类疾病等。且其生产过程引入的有机杂质、基因毒性杂质、无机杂质或残留的有机溶剂等均对人体健康有一定危害。因此,建立奥美拉唑引入杂质或残留有机溶剂的质量检测方法是十分有必要的,严格控制质量标准,把控药品市场质量安全。[b]1 仪器与材料1.1 实验仪器[/b]高效液相色谱仪(THERMO SCIENYIFIC, Mltimate3000);电子天平(METTLER-TOLEDO、BP-210S) [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](Agilent 6890N)顶空进样器(Agilent 7694E)[b] 1.2 实验试剂[/b][align=center][b]表1-1 实验所需试剂[/b][/align] [table][tr][td=2,1] [align=center][b]实验试剂[/b][/align] [/td][td] [align=center][b]厂家[/b][/align] [/td][/tr][tr][td=2,1] [align=center]磷酸二氢钾[/align] [/td][td] [align=center]莱阳经济技术开发区精细化工厂[/align] [/td][/tr][tr][td=2,1] [align=center]氢氧化钾[/align] [/td][td] [align=center]国药集团[/align] [/td][/tr][tr][td=2,1] [align=center]乙腈[/align] [/td][td] [align=center]Fisher Scientific[/align] [/td][/tr][tr][td=2,1] [align=center]4-甲氧基-2-硝基苯胺[/align] [/td][td] [align=center]北京百灵威科技有限公司[/align] [/td][/tr][tr][td=2,1] [align=center]4-甲氧基-邻苯二胺[/align] [/td][td] [align=center]Alfa Aesar[/align] [/td][/tr][tr][td=2,1] [align=center]奥美拉唑[/align] [/td][td] [align=center]寿光富康制药有限公司[/align] [/td][/tr][tr][td=1,6] [align=center]分析纯[/align] [/td][td] [align=center]丙酮[/align] [/td][td] [align=center]西陇化工股份有限公司[/align] [/td][/tr][tr][td] [align=center]甲醇[/align] [/td][td] [align=center]Fisher Scientific[/align] [/td][/tr][tr][td] [align=center]苯[/align] [/td][td] [align=center]天津富宇化工有限公司[/align] [/td][/tr][tr][td] [align=center]甲苯[/align] [/td][td] [align=center]莱阳经济技术开发区精细化工厂[/align] [/td][/tr][tr][td] [align=center]二氯甲烷[/align] [/td][td] [align=center]天津科密欧化学试剂有限公司[/align] [/td][/tr][tr][td] [align=center]DMA[/align] [/td][td] [align=center]Sigma-Aldrich[/align] [/td][/tr][/table][b]2 基因毒性杂质的检验方法的设定及方法学验证[/b]来源于起始物料苯并咪唑的合成路线的基因毒性杂质[sup][/sup]不适用于药典各论方法检测此类物质,在药典规定的波长无吸收。因此,采用液相色谱方法,对奥美拉唑成品中的4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺进行限度检测和控制。[b]2.1色谱条件[/b]色谱柱:ODS-3,5μm,4.6×250mm;检测波长分别设定为4-甲氧基-2-硝基苯胺(230nm)及4-甲氧基-邻苯二胺(210nm);流速为1.0ml/min;进样量为80μl;柱温为30℃。[b]2.2 溶液配制[/b]1) 流动相:溶解6.8g的磷酸二氢钾用纯化水溶解并稀释至1000ml,用氢氧化钾调节pH至6.5,和乙腈按(73:27)混合。2) 对照溶液:取4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺各16mg,精密称定置于200ml容量瓶中,用流动相溶解并稀释至刻度,准确量取1ml此溶液用流动相稀释至100ml,再量取1ml用流动相稀释至50ml。3) 奥美拉唑供试液:称取奥美拉唑样品100mg,精密称定置于50ml容量瓶中,用流动相溶解并稀释至刻度。注:计算奥美拉唑中的4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺含量都不得超过8ppm。[b]2.3质量检测方法验证[/b]通过限度验证,即该方法的专属性、系统适应性、检测限以及样品测定,是否符合验证可接受的标准,来判断该方法是否符合标准,可用于杂质测定。[b]2.3.1 专属性[/b]1) 溶液配制定性溶液:取4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺各16mg,精密称定置于200ml容量瓶中,用配制完毕的流动相溶解并稀释至刻度,准确量取1ml此溶液用流动相稀释至100ml,再稀释1ml用流动相稀释至50ml。2) 测定取流动相作为空白、定性溶液进样,记录色谱图,数据和结果。3) 数据与结果[align=center][b]表2-1专属性测试数据和结果[/b][/align] [table][tr][td] [align=center][b]项目[/b][/align] [/td][td] [align=center][b]4-甲氧基-2-硝基苯胺峰面积(230nm)[/b][/align] [/td][td] [align=center][b]4-甲氧基-邻苯二胺峰面积(210nm)[/b][/align] [/td][/tr][tr][td] [align=center]空白[/align] [align=center]溶液[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][tr][td] [align=center]定性[/align] [align=center]溶液[/align] [/td][td] [align=center]8550[/align] [/td][td] [align=center]12258[/align] [/td][/tr][/table][align=center][b]表2-2信噪比测试数据和结果[/b][/align] [table=100%][tr][td] [align=center][b]杂质[/b][/align] [/td][td] [align=center][b]信噪比[/b][/align] [/td][/tr][tr][td] [align=center]4-甲氧基-2-硝基苯胺峰面积(230nm)[/align] [/td][td] [align=center]0[/align] [/td][/tr][tr][td] [align=center]4-甲氧基-邻苯二胺峰面积(210nm)[/align] [/td][td] [align=center]0[/align] [/td][/tr][/table][b]2.3.2系统适用性试验[/b]1) 溶液制备贮备液:取4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺各16mg,精密称定置于200ml容量瓶中,用流动相溶解并稀释至刻度,准确量取1ml此溶液用流动相稀释至100ml。杂质溶液:用流动相稀释1ml贮备液到50ml或用专属性定性溶液及图谱。分离度:称取埃索美拉唑镁或奥美拉唑镁样品100mg,精密称定置于50ml容量瓶中,用流动相溶解后准确加入1ml贮备液并用流动相稀释至刻度。2) 测定以方法规定的色谱条件,取杂质溶液、分离度溶液分别进样,记录色谱图,数据和结果。3) 数据与结果[align=center][b]表2-3 系统适用性性测试结果[/b][/align] [table=562][tr][td] [align=center][b]溶液[/b][/align] [align=center][b]名称[/b][/align] [/td][td] [align=center][b]峰面积1[/b][/align] [/td][td] [align=center][b]峰面积2[/b][/align] [/td][td] [align=center][b]峰面积3[/b][/align] [/td][td] [align=center][b]峰面积4[/b][/align] [/td][td] [align=center][b]峰面积5[/b][/align] [/td][td] [align=center][b]峰面积6[/b][/align] [/td][td] [align=center][b]峰面积平均值[/b][/align] [/td][td] [align=center][b]RSD[/b][/align] [/td][/tr][tr][td] [align=center]4-甲氧基-2-硝基苯胺峰面积(230nm)[/align] [/td][td] [align=center]8427[/align] [/td][td] [align=center]8425[/align] [/td][td] [align=center]8481[/align] [/td][td] [align=center]8533[/align] [/td][td] [align=center]8483[/align] [/td][td] [align=center]8460[/align] [/td][td] [align=center]8468.17[/align] [/td][td] [align=center]0.48%[/align] [/td][/tr][tr][td] [align=center]4-甲氧基-邻苯二胺峰面积(210nm)[/align] [/td][td] [align=center]11701[/align] [/td][td] [align=center]11539[/align] [/td][td] [align=center]11086[/align] [/td][td] [align=center]11043[/align] [/td][td] [align=center]10548[/align] [/td][td] [align=center]10679[/align] [/td][td] [align=center]11099.33[/align] [/td][td] [align=center]4.11%[/align] [/td][/tr][/table][align=center][b]表2-4 奥美拉唑和4-甲氧基-邻苯二胺分离度测试结果[/b][/align] [table=100%][tr][td] [align=center][b]名称[/b][/align] [/td][td] [align=center][b]保留时间[/b][/align] [/td][td] [align=center][b]分离度[/b][/align] [/td][/tr][tr][td] [align=center]相邻杂质峰[/align] [/td][td] [align=center]3.813[/align] [/td][td] [align=center]-[/align] [/td][/tr][tr][td] [align=center]4-甲氧基-邻苯二胺峰面积(210nm)[/align] [/td][td] [align=center]4.736[/align] [/td][td] [align=center]2.16[/align] [/td][/tr][tr][td] [align=center]相邻杂质峰[/align] [/td][td] [align=center]5.248[/align] [/td][td] [align=center]1.69[/align] [/td][/tr][/table][align=center][b]表 2-5奥美拉唑和4-甲氧基-2-硝基苯胺分离度测试结果[/b][/align] [table=100%][tr][td] [align=center][b]名称[/b][/align] [/td][td] [align=center][b]保留时间[/b][/align] [/td][td] [align=center][b]分离度[/b][/align] [/td][/tr][tr][td] [align=center]相邻杂质峰[/align] [/td][td] [align=center]23.168[/align] [/td][td] [align=center]-[/align] [/td][/tr][tr][td] [align=center]4-甲氧基-2-硝基苯胺峰面积(230nm)[/align] [/td][td] [align=center]26.908[/align] [/td][td] [align=center]3.32[/align] [/td][/tr][tr][td] [align=center]相邻杂质峰[/align] [/td][td] [align=center]29.467[/align] [/td][td] [align=center]2.85[/align] [/td][/tr][/table][b]2.3.3检测限[/b]1) 溶液制备按照选择项下贮备溶液的配制方法配制溶液,并将标准溶液逐步稀释,得到适当浓度的溶液。2) 测定在色谱条件下,取溶液进样,记录色谱图。当待测组分的信噪比大于2时,对应的浓度为该组分的最小检测浓度。3) 数据与结果4-甲氧基-2-硝基苯胺检测限0.00256 μg/ml,LOD=1.28ppm,S/N=2.22 4-甲氧基-邻苯二胺检测限0.00256μg/ml,LOD=0.000128,S/N=2.[b]2.3.4样品检测[/b]1) 溶液配制根据已设定检测方法已将溶液配制完毕。2) 测定分别取三批样品按照溶液的配制方法,配制供试液进样,记录色谱图。3) 数据与结果[align=center][b]表2-6 奥美拉唑样品检测结果[/b][/align] [table=102%][tr][td] [align=center][b]批号[/b][/align] [/td][td] [align=center][b]4-甲氧基-2-硝基苯胺(230nm)[/b][/align] [/td][td] [align=center][b]4-甲氧基-邻苯二胺(210nm)[/b][/align] [/td][/tr][tr][td] [align=center]20150401[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][tr][td] [align=center]20150402[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][tr][td] [align=center]20150403[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][/table][b]3残留溶剂的检测方法的设定[/b]在《中国药典》[sup][/sup]规定的奥美拉唑中各论残留溶剂的检测方法的基础上,进行修正,更改部分参数,选用内标法对残留溶剂进行检测,有利于快速检验及产品及时入库。[b]3.1 色谱条件[/b]1) [url=https://insevent.instrument.com.cn/t/Mp]气相[/url]部分色谱柱:Agilent DB-624, 0.32mm×30m,膜厚1.8μm;柱温先以50 ℃保持5分钟,后以20℃/min升温到200℃保持4分钟;进样口温度为200℃; 分流比为1:1;检测器为FID,其温度为300℃;载气设定为氮气;柱流量则为3.0ml/min。2) 顶空部分顶空瓶平衡温度98℃,平衡时间20min;定量环温度115℃,体积1ml;传输管线温度为130℃。[b]3.2 溶液配制[/b]1) 苯贮备液:精密称取苯0.02g于已加入少量DMA的100ml容量瓶中,用DMA稀释至刻度,摇匀。2) 标准贮备液:精密称取丙酮0.15g,甲醇0.1g,二氯甲烷0.01g,甲苯0.03g,于已加入少量DMA的100ml容量瓶中,在此容量瓶中加入1ml准确量取的苯贮备液,用DMA稀释至刻度,摇匀。3) 标准溶液:精密量取标准溶液贮备液5.0ml于50 ml容量瓶中,用DMA稀释至刻度,混合均匀。4) 供试溶液:精密称定样品0.5g于20ml顶空瓶中,用5ml DMA溶解。[b]3.3 检测方法[/b]1) 按照[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]部分和顶空部分的操作条件设定操作方法。取标准溶液顶空进样,记录色谱图(主要组分出峰顺序依次为甲醇、丙酮、二氯甲烷、苯、甲苯)。注:计算相邻组分之间的分离度R,均应不小于1.5;取6份标准溶液,连续进样,计算各溶剂峰面积的RSD,应不大于10%。2) 先将空白溶液、6份标准溶液和样品溶液各5ml置于顶空瓶中,密封。取空白溶液进样,记录图谱,再取6份标准溶液,记录色谱图,进行系统适用性试验和标准校正,最后取供试溶液进样,记录图谱。计算公式如下式(2-1):[align=center][img=,211,60]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241130224283_2738_3389662_3.png!w211x60.jpg[/img];[/align][align=center][img=,187,81]https://ng1.17img.cn/bbsfiles/images/2019/07/201907241130597462_639_3389662_3.png!w187x81.jpg[/img][/align]注:X[sub]1[/sub]:残留甲醇、甲苯、二氯甲烷、丙酮的量,ppm X[sub]2[/sub]:残留苯的量,ppm Ai:供试溶液的图谱中溶剂(i)的峰面积;A[sub]0[/sub]:空白溶液的图谱中溶剂(i)的峰面积;A[sub]si[/sub]:标准溶液的图谱中溶剂(i)的峰面积;W:样品的称量,g;W[sub]si[/sub]:溶剂(i)的称重,g。[b]3.4 检测结果[/b][align=center][b]表3-1 奥美拉唑残留溶剂检验结果[/b][/align] [table][tr][td=1,2] [align=center][b]项目[/b][/align] [/td][td=1,2] [align=center][b]标准[/b][/align] [/td][td=1,2] [align=center][b]方法[/b][/align] [/td][td=3,1] [align=center][b]奥美拉唑检验批号[/b][/align] [/td][/tr][tr][td] [align=center][b]A-51511507002[/b][/align] [/td][td] [align=center][b]A-51511507003[/b][/align] [/td][td] [align=center][b]A-51511507004[/b][/align] [/td][/tr][tr][td=1,5] [align=center]残留溶剂检验[/align] [/td][td] [align=center]丙酮不得超过1500ppm[/align] [/td][td=1,5] [align=center]内控[/align] [/td][td] [align=center]309ppm[/align] [/td][td] [align=center]396ppm[/align] [/td][td] [align=center]423ppm[/align] [/td][/tr][tr][td] [align=center]二氯甲烷不得超过100ppm[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][tr][td] [align=center]甲醇不得超过500ppm[/align] [/td][td] [align=center]115ppm[/align] [/td][td] [align=center]129ppm[/align] [/td][td] [align=center]122ppm[/align] [/td][/tr][tr][td] [align=center]甲苯不得超过300ppm[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][tr][td] [align=center]苯不得超过1ppm(LOQ)[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][td] [align=center]未检出[/align] [/td][/tr][/table][b]4小结[/b]本研究对治疗胃溃疡的一线药物奥美拉唑进行质量检验方法的研究。通过分析其合成过程中引入的杂质,创新性的提出原料药中可能存在的基因毒性杂质4-甲氧基-2-硝基苯胺、4-甲氧基-邻苯二胺,同时对生产过程引入的残留有机溶剂进行质量监控。根据ICH的指南Q2A和Q2B的要求,采用液相色谱,对奥美拉唑成品中的4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺进行限度检测,并对检测方法进行了专属性、系统适应性、检测限,样品测定等方面的限度验证。限度验证结果均应符合标准,说明该检测方法符合测定的准确性、可靠性和灵敏度的要求,能够进行该杂质的测定。且使用该方法进行三种批号的奥美拉唑基因毒性杂质检验时,均未发现存在4-甲氧基-2-硝基苯胺和4-甲氧基-邻苯二胺。说明现有的工艺可有效除去原料药中引入的这两种基因毒性杂质,可不放入日常质量监控之中。同时,在对奥美拉唑合成工艺中残留的有机溶剂的质量检测研究中,进行检测时,发现,其药品中检测出少量的丙酮和甲醇,但均在质量标准规定以内,未检测出二氯甲烷、甲苯、苯,说明选用的三批奥美拉唑成品药均符合药品质量标准。而在检测中,本研究创新性使用不同于中华人民共和国药典中的N ,N-二甲基甲酰胺(DMF),而选择易于冲洗的N ,N-二甲基乙酰胺(DMA)做溶媒,易冲洗干净,且不影响公司内其它产品的检测,与中华人民共和国药典方法相比,大大缩短检验样品的时间,中华人民共和国药典方法单个样品的检测时间为65min,内控的方法仅为36.5min,对工业化规模生产来说,快速检测样品既经济又能保证产品及时入库。[b]参考文献[/b] AnaLuisa Correia, Mina J Bissell. The tumor microenvironment is a dominantforceinmulti drμg resistance.Drμg Resist Update. 2012, 15(6):39-49. Shaojun Shi, ΜlrichKlotz,Protonpump inhibitors: an update of their clinical us and pharmacokinetics .EurJ Clin Pharmacol, 2008, 64(30): 935-951. ICHVALIDATION OF ANALYTICAL PROCEDURES: TEXT AND METHODOLOGY Q2 (R1) Current Step4 version (Complementary Guideline on Methodology dated 6 November 1996incorporated in November 2005). 国家药典委员会.中华人民共和国药典.二部.北京:中国医药科技出版社, 2015: 1412.
他达拉非杂质是一种化学物质,它是他达拉非的同分异构体或相关化合物。他达拉非是一种磷酸酯酶抑制剂,用于治疗男性勃起功能障碍。COTO标准品是一种高纯度的标准物质,用于测定他达拉非及其杂质的纯度、含量和化学性质。通过与COTO标准品进行对比和分析,可以确定他达拉非及其杂质的结构、组成和含量,从而保证他达拉非的质量和安全性。在药物研发和生产过程中,COTO标准品的使用非常重要。它可以提供可靠的参照物,用于质量控制、药物分析和化学计量学研究。通过使用COTO标准品,可以确保他达拉非及其杂质的准确性和可靠性,为药物的安全性和有效性提供保障。总的来说,COTO标准品在他达拉非杂质的研究和控制中具有重要作用。通过使用COTO标准品,可以更好地了解他达拉非及其杂质的性质和含量,从而确保药物的安全和有效性。同时,也需要加强生产过程中的管理和监督,加强质量标准和监管措施的执行力度,确保药物质量和安全。
如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
艾普拉唑杂质是一种化学物质,它是艾普拉唑的同分异构体或相关化合物。艾普拉唑是一种质子泵抑制剂,用于治疗胃溃疡、十二指肠溃疡和反流性食管炎等疾病。COTO标准品是一种高纯度的标准物质,用于测定艾普拉唑及其杂质的纯度、含量和化学性质。通过与COTO标准品进行对比和分析,可以确定艾普拉唑及其杂质的结构、组成和含量,从而保证艾普拉唑的质量和安全性。在药物研发和生产过程中,COTO标准品的使用非常重要。它可以提供可靠的参照物,用于质量控制、药物分析和化学计量学研究。通过使用COTO标准品,可以确保艾普拉唑及其杂质的准确性和可靠性,为药物的安全性和有效性提供保障。总的来说,COTO标准品在艾普拉唑杂质的研究和控制中具有重要作用。通过使用COTO标准品,可以更好地了解艾普拉唑及其杂质的性质和含量,从而确保药物的安全和有效性。同时,也需要加强生产过程中的管理和监督,加强质量标准和监管措施的执行力度,确保药物质量和安全。
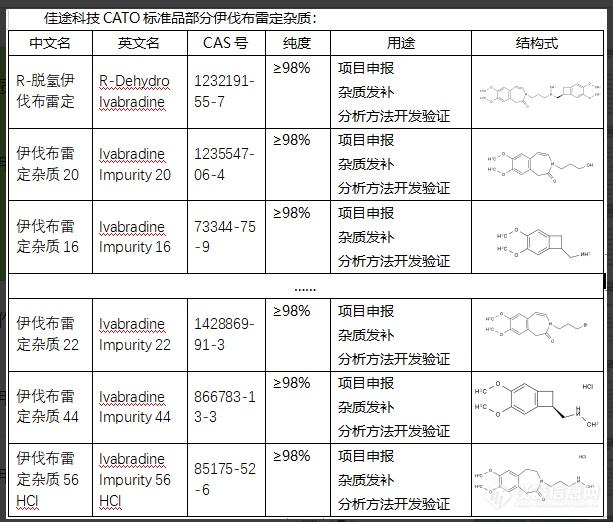
伊伐布雷定是一种用于治疗成人和儿童音乐综合症等疾病的药物。在伊伐布雷定的制造过程中,可能会产生一些杂质。这些伊伐布雷定杂质可能由化学反应生成,也可能来自原始材料。无论杂质来源如何,过多的杂质可能会影响药品的质量、疗效和安全性。例如,一些杂质可能会导致药物的疗效降低,或者引发不良反应。因此,制药公司必须在生产过程中严格检测和控制这些杂质,以确保药品的质量和安全性。CATO标准品对杂质进行研究和分析也可以有助于优化制药过程,例如,找出产生杂质的环节并进行调整,以减少杂质的生成。这有助于提高药品质量,保证疗效,保证患者的安全。[img=,613,522]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052104420028_3541_6381668_3.png!w613x522.jpg[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP