我想了解4-氯-2-三氟甲基苯腈(4-Chloro-2-(trifluoromethyl)benzonitrile,CAS#320-41-2)的理化性质,但在网上只找到沸点109 º C (10 mmHg),是液体还是固体看不出来。因为这个沸点是真空条件下的。那位老师有相关的信息,请告诉我,谢谢!
我们做对氟苯甲酸检测,别人推荐用N,O-双(三甲基硅基)乙酰胺反应后测他们的衍生物,可是我搞不明白这个反应原理,不知道是不是可以反应彻底,请诸位高手帮忙讲解一下,谢谢!
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
联苯苄基氯(学名;4,4’-双氯甲基联苯)溶解方法?用ICP-AES测定其中的Ca,Fe,Zn,P,Na等元素(1--5ppm)。请高手指点样品处理方法为盼。谢谢啊http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
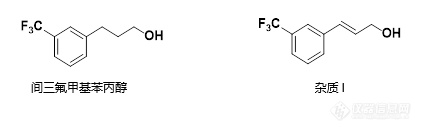
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
苯环上3位和5位上各带一个三氟甲基,扫碳谱后非常的杂,如何才能比较准确地找出相对应的峰并计算耦合常数呢?三氟甲基上的碳是否裂分位一个4重峰了,临位的碳也被裂分为双峰了呢?
有的书上说,三氟甲基在碳谱中可产生1:3:3:1的四重峰,距离比较远。本人做过一次,基本上只得到两个较高峰,两边的小峰有扫不出来的可能吗?而且,三氟甲基对alfa碳有裂分吗?
[size=4][font=宋体]求香菇上氟乐灵,甲基毒死蜱,毒死蜱,氯杀螨,乙硫磷,氯苯嘧啶醇[/font][/size][size=4][font=Times New Roman] [/font][/size][size=4][font=宋体]氟虫腈的前处理方法,能够有效去除杂质的,谢谢大家,这个香菇上的杂质太多了,用弗洛里硅土不管用,杂质很多。[/font][/size]
三甲基氢醌与三甲基苯醌之间的关系,怎么计算两者之间的含量
[size=18px]目前在用AB的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测三苯基氯甲烷,Q1 MI模式扫243.1的离子[font=-apple-system, BlinkMacSystemFont, &](应该是三苯甲基碳正离子)[/font],发现基线非常高(30万-50万之间),且不稳定,时高时低,导致峰面积也 不稳定,打电话问客服,几个人几种说法,“液相部分污染了”“这个是正常现象,多走走就稳定了”,尝试用MRM模式去做,打出一个165.2的碎片,基线不到1000,做了线性和回收也都挺好,但是,这个碎片离子是怎么打出来的比较困惑,就怕以后再做的时候重现不出来……[/size][size=18px]流动相是90%甲醇,溶剂是正丁醇:乙腈(80:20)[/size][size=18px]请教一下各位大神,AB的仪器用SIM模式选择Q1 MI还是Q3 MI好呢?基线高且时高时低,除了污染还有什么原因呢?[font=-apple-system, BlinkMacSystemFont, &]三苯甲基碳正离子在质谱里能被打碎吗?会裂解成什么碎片离子?[/font][/size][size=18px][font=-apple-system, BlinkMacSystemFont, &][/font][/size]
液相色谱峰刚开始有正峰和反峰连在一起,乙腈和水做流动相的,是正常的吗,分析的2.4.6-三甲基苯甲酰氯含量甲醇酯化做的
我们分析2.4.6-三甲基苯甲酰氯,我的岛津液相出峰开始会出个倒峰是什么原因,紫外 C18 乙腈水 1ml/min
我用滴定对苯醌的方法(溶液加碘化钾,盐酸,暗处静置后用硫代硫酸钠滴定)测三甲基苯醌含量,但是终点总是反色,找不到终点,请问高人们有解决的办法吗?谢谢!
PONY谱尼测试最新了解到,厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。PONY谱尼测试在农药残留方面具有丰富的经验,可以依据日本肯定列表进行检测。对于这三种农药,企业应该引起重视,PONY谱尼测试将鼎力帮助企业进行检测,顺利出口。
请问谁有以下标准可否给我一份,谢谢了甲基硫菌灵的标准HG 2462.2-1993福美双的标准HG3758-2004
有什么比较好的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]方法定量检测N,O-双(三甲基硅基)乙酰胺中乙酰胺。用什么试剂做溶剂能够溶解乙酰胺同时又不与N,O-双(三甲基硅基)乙酰胺反应呢?
各位老师 我想 求一下 甲基肼, 三氯化磷,磷化氢的标准曲线和相对应的吸光度 谢谢 甲基肼-对二甲胺基苯甲醛分光光度法 三氯化磷-对氨基二甲基苯胺分光光度法 磷化氢-钼酸铵分光光度法 五氧化二磷-钼酸铵分光光度法谢谢各位老师
厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。
我用的是250mm的C18柱,紫外检测器,检测波长243和280nm,流动相:甲醇和10%乙酸溶液,梯度洗脱,柱温40℃,流速0.8mL/min,进样量:20μ,测藜芦醛和N-甲基甲酰苯胺,这俩峰咋分不开,是哪里的问题啊,恳请各位大佬指教
如题,双氯甲醚与2,4,6-三氯苯酚钠盐在一定条件下反应生成什么呢?(已知氯甲醚与2,4,6-三氯苯酚钠盐在一定条件下反应生成a-甲氧基三氯苯甲醚)
各位仁兄:三氟甲基磺酸含量如何测定?
含有三氟甲基的化合物其碳谱裂分有什么规律吗?
标题:出处:安徽大学学报:自然科学版 1999年23卷3期作者:聂康明[1] 李蕤[2] [1]安徽大学化学系 [2]合肥联合大学化工系 摘要:以甲基丙烯酸和二缩三乙二醇为原料,合成用于制备PP/PnBA IPN共混材料的交联剂--双内烯酸二缩三乙二醇酯,应用傅里叶红谚氢核磁共振谱(H-NMR)表征了产物的化学结构,分析表明,填充互穿聚合物组合组分的相容性交联剂的化学结构有关,长链柔顺性罗好的双人烯酸二缩三乙二醇酯使填充IPN共混体系双组分的Tg值内移程度较大,表现出较好的强迫增容效果。
GB/T29668-2013化妆品用防腐剂 双(羟甲基)咪唑烷基脲
最近在做国标21169-2007蜂蜜中双甲脒及其代谢物的扩项(液相色谱法),按照方法做出来,双甲脒回收率在百分之七八十左右,2,4-二甲基苯胺在百分之三四十左右,死活都达不到标准方法里说的双甲脒回收率99.8%以上,2,4-二甲基苯胺71.7%以上。不知道有没有老师做过这个方法,怎么才能提高双甲脒和2,4-二甲基苯胺的回收率?
谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法
本人做蜂蜜中双甲脒及其代谢物,双甲脒和2,4二甲基苯胺是混标,采用方法农业部781-2008,回收双甲脒很好,但是2,4二甲基苯胺很低,重复实验也是如此,标曲线性很好,请问可能的原因是什么?
1. 在用亚甲蓝测水中硫化物的时候,需要配置0.2%的对氨基二甲基苯胺溶液,我查了一下,他的别名也可以叫叫[font=宋体, Arial, Helvetica, sans-serif][size=12px]N,N-二甲基对苯二胺二盐酸盐,应该没买错吧。[/size][/font]2. 他的储存条件不是-20℃吗?这样它里面会不会含有很高的水分?那我称取2 g的 时候,需不需要干燥,还是直接就称取2 g 就好了。3. 还有就是配置好0.2%的对氨基二甲基苯胺溶液后,怎么储存呢?室温?冷藏?冷冻?如有回复,万分感谢!![img=,690,449]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239193272_1018_5383916_3.png!w690x449.jpg[/img][img=,690,299]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239240158_2694_5383916_3.png!w690x299.jpg[/img]
亚甲基双水杨酸杆菌肽混剂(效价)的测定 用液相色谱法 我做不出峰 没有相关方法
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]测有机酸,色谱柱是DB-5MS,是非极性的图层,需要用到 N,O-双(三甲基硅基)三氟乙酰胺(1%三甲基氯硅烷),这个试剂含水吗?会损伤柱子吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP