请问除了丙酮,氩气可以进行手动捡漏时候来观察离子变化外,还有什么化合物可以呢?
气相色谱法测定环境空气中硝基苯类化合物,如果没有色谱纯的丙酮做溶剂,可以用分析纯的丙酮代替吗?
甲醛测定的测定方法:乙酰丙酮法原理是利用甲醛与乙酰丙酮及氨生成黄色化合物二乙酰基二氢卢剔啶后,412nm下进行分光光度测定。此法最大的优点是操作简便,性能稳定,误差小,不受乙醛的干扰,有色溶液可稳定存在12hr,;缺点是灵敏度较低,最低检出浓度为0.25mg/L,仅适用于较高浓度甲醛的测定;方法缺点是反应较慢,需要约60min;SO2对测定存在干扰(使用NaHSO3作为保护剂则可以消除)。
不同国家地区的分法:美国职业卫生研究所1973年登记的有毒化学物质已达25043种,主要化和毒物可分为: 重金属如Hg,Pb,As,Cd,Cr等。 有机物如有机氯农药,多环芳烃,多氯联苯,氯代苯,亚硝胺类,有机汞等。 欧洲共同体在1975年根据物质的毒性,持久性和生物积累性列出了有害有毒物质的“黑名单”,“黑名单”中不包括那些生物学上无害的物质和易转化为生物学上无害的物质。 1.有机卤化物和可以在环境中形成卤化物的物质 2.有机磷化合物 3.有机锡化合物 4.在水环境中或由于水环境介入而显示致癌治性的物质 5.汞及其化合物 6.镉及其化合物 7.持久性油类和来自石油的烃类 8.可漂浮、悬浮或下沉和妨碍水质的任何持久性物质联邦德国在1980年公布了120种水中有害物质名单,其中毒性最强的有16种,它们是;丙酮氰醇,丙烯腈,砷酸氢二钠,苯,四乙基铅,镉化合物,氰化物,DDT,3-氯环氧丙烷-l,2,乙烯亚胺,水合肼,林丹,硫醇,乙基对硫磷,汞化合物,银化合物。
参照行标测试印刷制品中的VOCS,其中有一项为丙酮,选用的目标离子是43,特征离子为45,58,保留时间6.584min.但我在实际操作中发现,实际样品的匹配度(计算报告中的偏差)只有7,而其他几种测试挥发性物质的匹配度至少都80以上。对比了质谱图,丰度比类似的只有一个叫二甲基二氮烯的化合物。进一步进行NTST搜索,通过58,43两个离子丰度比的比较,能基本判断是丙酮。但我想不明白的是,为啥匹配度这么低也能对上啊,想问下有没同仁碰到过类似问题的?
求分离一氯丙酮、1,3-二氯丙酮、1,1-二氯丙酮、1,1,3-三氯丙酮、1,1,1-三氯丙酮、1,1,1,3-四氯丙酮的GC分析方法?
下面属于闭链化合物的是()。 A、甲烷 B、丙酮 C、乙醚 D、甲苯
[color=black]天然橡胶([/color][color=black]NR[/color][color=black])是从天然植物中获取的以聚顺式[/color][color=black]1,4[/color][color=black]异戊二烯为主要成分的天然高分子化合物,除了橡胶烃外,还含有少量的非胶物质。[/color]与合成橡胶相比,天然橡胶具有更加优异的综合性能,究其原因,除了与橡胶的分子量及分子量分布有关外,橡胶中所含的非胶组分及含量也对其综合性能产生着很大影响。[color=black]其中[/color]天然橡胶中所有能溶于丙酮的[color=black]高级脂肪酸及固醇类[/color]物质称为丙酮溶解物,其主要成分为硬脂酸、油酸、亚油酸、甾醇及甾醇脂,另外还有少量的α-生育酚和γ、α和δ-三烯生育酚等天然防老剂。一般认为丙酮溶解物对橡胶起硫化活性剂、脱模剂、抗氧化剂等多种作用。例如脂肪酸盐类同样在合成橡胶(除了丁基橡胶)、天然橡胶及天然胶乳中有所应用。不仅作为活性剂,在耐磨材料中可作增塑剂,在硫化率差异大的胶料中可以作稳定剂。硬脂酸锌、硬脂酸钡和硬脂酸钙等在塑料中作为光稳定剂和热稳定剂使用,也可以作为塑料的脱模剂。醇类和固醇类活性剂一方面可以用于炭黑补强的合成橡胶、天然橡胶及胶乳,另一方面在含有白炭黑的胶料中起到活化作用,同时具有防水功能,并在稳定胶料硬度方面表现出出色的效果。
很多种类的极性化合物分离条件。 UDP-葡萄糖 UDP-葡萄糖、UDP-半乳糖、磷酸半乳糖 葡萄糖 蔗糖 红细胞中的UDP-葡萄糖、UDP-半乳糖、三磷酸腺苷(ATP) ADP-葡萄糖、CDP-葡萄糖 糖核苷酸 胞嘧啶、胸腺嘧啶、尿嘧啶、鸟嘌呤、腺嘌呤 三磷酸腺苷(ATP)、一磷酸腺苷(AMP) 黄嘌呤-磷酸、鸟嘌呤-三磷酸 体液中的黄嘌呤、尿酸、次黄嘌呤 色胺、五羟色胺、多巴胺 L-天冬氨酸、L-精氨酸 L-精氨酸、L-赖氨酸、L-组氨酸 谷氨酸、赖氨酸 亮氨酸、异亮氨酸 L-甲硫氨酸、L-谷氨酸 甲基琥珀酸、戊二酸、草酸、肌酸、4-羟脯氨酸、天冬氨酸、鸟氨酸 叶酸 抗坏血酸 胆汁酸 柠檬酸、马来酸、反式乌头酸 马来酸、富马酸 3-羟基肉桂酸 矮壮素、甲哌啶 苯海拉明 4-二甲氨基吡啶 草甘膦 三聚氰胺、三聚氰酸 胍
很多种类的极性化合物分离条件。 UDP-葡萄糖 UDP-葡萄糖、UDP-半乳糖、磷酸半乳糖 葡萄糖 蔗糖 红细胞中的UDP-葡萄糖、UDP-半乳糖、三磷酸腺苷(ATP) ADP-葡萄糖、CDP-葡萄糖 糖核苷酸 胞嘧啶、胸腺嘧啶、尿嘧啶、鸟嘌呤、腺嘌呤 三磷酸腺苷(ATP)、一磷酸腺苷(AMP) 黄嘌呤-磷酸、鸟嘌呤-三磷酸 体液中的黄嘌呤、尿酸、次黄嘌呤 色胺、五羟色胺、多巴胺 L-天冬氨酸、L-精氨酸 L-精氨酸、L-赖氨酸、L-组氨酸 谷氨酸、赖氨酸 亮氨酸、异亮氨酸 L-甲硫氨酸、L-谷氨酸 甲基琥珀酸、戊二酸、草酸、肌酸、4-羟脯氨酸、天冬氨酸、鸟氨酸 叶酸 抗坏血酸 胆汁酸 柠檬酸、马来酸、反式乌头酸 马来酸、富马酸 3-羟基肉桂酸 矮壮素、甲哌啶 苯海拉明 4-二甲氨基吡啶 草甘膦 三聚氰胺、三聚氰酸 胍
丙酮是非极性化合物,但它为什么易溶于极性的水呢?
丙酮的理化性质、毒性及安全防护 丙酮 CH3COCH3 1、别名英文名 二甲基甲酮、阿西通、醋酮;Acetone、Dimethyl ketone、Propanone。 2、用途 用于有机溶剂、醋酐、氯仿、染料、石腊的精炼、橡胶、润滑油的制造。 3、制法 (1)通过玉米发酵法分馏制得。 (2)石油裂解法制得。 (3)木材干馏。 (4)醋酸钙加热。 (5)异丙醉催化脱氢。 4、现化性质 分子量: 58.08 熔点: 一94.6℃ 沸点: 56.48℃ 液体密度(15℃): 797.2kg/m3 气体密度: 2.00kg/m3 压缩系数(14.2℃,90L 79~3699.38kPa):0.00011l 临界温度: 236.5℃ 临界压力: 4782.54kPa 临界密度: 278kg/m3 气化热(0℃): 563.79kJ/kg 比热容(气体,26~110℃,101.325kPa): Cp=1452.37J/(kgK) 蒸气压(39.5℃): 53.33kPa (25℃): 30.17kPa 粘度(气体,0℃): 0.00725mPas (液体,0℃): 0.4013mPas 表面张力(丙酮一空气或蒸气,0℃): 26.2mN/m 导热系数(0℃,气体): 0.0096338W/(mK) (0℃,液体): 0.177702W/(mK) 折射率: 1.3585 闪点: 一17.78℃ 燃点: 465℃ 爆炸界限: 2.6%一12.8% 最大爆炸压力: 872。79kPa 产生最大爆炸压力的浓度: 6.3% 最易引燃浓度: 4.5 燃烧热(液体,25℃): 1791. 62kJ/mo1 毒性级别: 1 易燃性级别: 3 易爆性级别: 0 丙酮在常温压下为具有特殊芳香气味的易挥发性无色透明液体。易燃烧,其蒸气空气能形成爆炸性混合物,遇明火或高热易引起燃烧。化学性质较活泼。其液体比水轻。能与水、酒精、乙醚、氯仿、乙炔、油类及碳氢化合物相互溶解,能溶解油脂和橡胶。丙酮蒸气有麻醉效应。 丙酮与一些物质混合接触时的危险性如下表所示。 5、毒性 兔一经口 LD50:5300mg/㎏ 最高容许浓度:1000ppm(2400mg/㎏) 丙酮可经呼吸道、消化道和皮肤吸收。经皮肤吸收缓慢,量少,主要是驼过前两个途径。由于丙酮的水溶性强,它易溶解和吸收入血液中,很快分布于全身。丙酮属微毒类,其毒性主要是对中枢神经系统的麻醉作用。基蒸气对粘膜有中等程度的刺激作用。丙酮对皮肤无致敏作用,但有轻度刺激作用。 吸信蒸气急性中毒后主要表现为不同程度的麻醉状态。最初出现管乏力、恶心、头痛、头晕、容易激动。严重时可出现呕吐、气急、痉挛以及昏迷。液体能刺激眼睛。吞服反能刺激消化系统,产生麻醉与错迷等症状。 6、安全防护 丙酮要用玻璃羡慕或金属桶盛装。灌装时控制流速不可过快,防止产生静电。库内存放丙酮时,不应把库房堆满,在墙边,离顶栅、离柱子应留出一定的通道,以便进行防火检查及发生火灾时有效地进行消防施救。 发生火灾时可用抗溶性泡沫、二氧化碳、化学干粉、黄砂灭火。用水灭火可能无效,但可用来冷却火场中的容器,驱散蒸气,稀释和赶走流出的液体,保护去堵漏的人员。 丙酮流出时,首先要切断所有火原,穿戴好防毒面具、手套防护服等,用水冲洗,洗水经稀释后排入废水系统。
我在做生物胺的实验 其中需要配10mg/mL的丹磺酰氯丙酮溶液 最后发现不溶 丹磺酰氯是新买新开封的 丙酮也开了瓶新的 超声效果也不理想 这是什么原因 有人能帮帮我吗

乙酰丙酮分光光度法测定水中甲醛的不确定度分析1.方法原理及操作流程1.1原理在过量氨盐存在下,甲醛与乙酰丙酮生成黄色化合物,该有色化合物在414nm波长处有最大吸收。1.2操作流程量取100ml试样,经预蒸馏除去干扰物质;用100ml容量瓶接受馏出液并定容。用25ml具塞比色管量取25ml试样,加入2.5ml乙酰丙酮溶液,摇匀。于45-60℃水浴中加热30min,取出冷却。用10mm比色皿在波长413nm处,以水为参比测量吸光度。同时绘制校准曲线:取6支25ml具塞比色管,依次加入0.00、0.50、1.00、3.00、5.00、8.00ml 10.0ug/ml甲醛标准使用液,再分别加水至25ml。按样品测试步骤测定出标准系列的吸光度。由校准系列所测得的吸光度减去零管的吸光度值,绘制吸光度-甲醛含量的曲线;根据测得得试样中甲醛的吸光度(试样的吸光度扣除零空白试验的吸光度)从校准曲线上查得试样中甲醛的质量(ug),再除以试份的体积(ml),即得到试样中甲醛的浓度。1.3甲醛标准使用液的配制直接购买有证标准物质37.25mg/L的甲醛标准,用15ml分度吸管准确吸取13.4ml甲醛标准溶液至50ml容量瓶中,用水稀释至刻度,摇匀,此溶液即为10.0ug/ml甲醛标准使用液。1.4样品测量通过校准曲线拟合,用乙酰丙酮分光光度法测量样品中甲醛的浓度。得数据:http://ng1.17img.cn/bbsfiles/images/2013/03/201303311454_433120_2139979_3.jpg其中u(C)—C的标准测量不确定度u(m)—m的标准测量不确定度 u(V)—V的标准测量不确定度3.测量m的标准不确定度分量测量m的标准测量不确定度分量由三部分构成,其一是由标准溶液的质量-吸光度拟合的直线求得m时所产生的不确定度,记为u1(m);其二是由甲醛标准溶液配制成不同浓度的标准溶液系列时所产生的测量不确定度,记为u2(m)。3.1 u1(m)的计算甲醛校准曲线方程表示为:y=bx+a (3)式中,x—溶液中甲醛的质量,ug y—甲醛质量为x时对应的吸光度 b—校准曲线的斜率,b=0.00996(本次样品考核数据) a—校准曲线的截距,a=-0.001(本次样品考核数据)http://ng1.17img.cn/bbsfiles/images/2013/03/201303311456_433121_2139979_3.jpg3.2 u2(m)的计算绘制校准曲线的标准系列,其甲醛的质量可用下式来表示:mi=C0×V标 (5)式中,C0—为甲醛标准使用液的浓度,10.0ug/mlV标—为标准曲线标准系列中某一浓度点对应的加入甲醛标准使用液的体积,mlmi—为校准曲线标准系列中某一浓度点对应的甲醛的质量,ug10.0ug/ml的甲醛标准使用液是由标准贮备液经过稀释得到,用公式表示为:C0=C贮/(f)(6)式中,C贮—为甲醛标准贮备液的准确浓度,ug/mlf—为稀释因子,f代表甲醛贮备液稀释倍数。直接购买37.25 ug/ml甲醛标准物质作为贮备液稀释得到10.0ug/ml甲醛使用液。标准溶液的稀释是采用15ml的分度吸管和50ml的容量瓶来完成。http://ng1.17img.cn/bbsfiles/images/2013/03/201303311457_433122_2139979_3.jpg式中,[font=Times New Roma
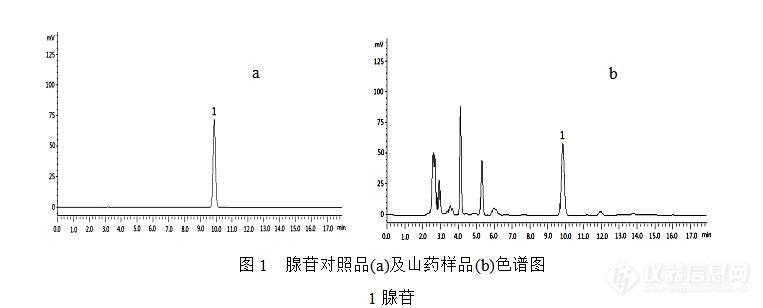
[align=center][b]HPLC测定不同来源山药中腺苷和脱氢表雄酮的含量[/b][/align] 山药(Dioscoreae Rhizoma)是薯蓣科植物薯蓣([i]Dioscorea opposita [/i]Thunb.)的干燥块茎,具有补脾养胃、生津益肺、补肾涩精的功能,是国家卫生部公布的药食两用的中药。然而在2015版《中国药典》一部中只收载了性状、鉴别、检查和浸出物等项目,缺少含量测定项指标,不足以对山药质量进行全面有效的控制。DHEA是人体肾上腺素的一种,国内外研究已经证实,DHEA对预防和治疗心血管疾病、增强免疫力、延缓衰老等均有一定效果,是山药中的重要活性成分之一。另外,腺苷具有舒张血管、降低血压、减慢心律、抑制血小板聚集、松弛血管平滑肌等生理活性。长期以来,由于对山药品种的评价和改良工作进展缓慢,目前仍然存在山药种植品种杂乱,缺乏综合性优良的品种。因此,本研究对包括野生山药在内的不同来源山药中的腺苷、DHEA含量进行测定,旨在为山药质量标准建立提供参考依据,也为山药品种驯化和选育提供一定的研究基础。[b][b]1 仪器和试药[/b][/b]LC-20AD高效液相色谱仪,配备紫外可见检测器(日本,岛津公司);ME204电子天平(德国Mettler Toledo公司),KQ-E型超声波清洗器(昆山市超声仪器有限公司)。脱氢表雄酮标准品(阿拉丁,批号为H1408213),腺苷标准品(中国食品药品检定研究院,批号为110879-200202);甲醇为色谱纯,水为娃哈哈纯净水,其他试剂均为分析纯。山药样品采自河南省温县山药种植基地。[b][b]2 方法与结果2.1标准储备溶液的制备[/b][/b]精密称取腺苷对照品6.61 mg置25 mL容量瓶中,加15 %甲醇溶解稀释至刻度,摇匀,配制成浓度为264.40 μgmL[sup]-[/sup][sup]1[/sup]的腺苷标准品储备溶液。精密称取DHEA对照品7.62 mg置50 mL容量瓶中,加甲醇溶解稀释至刻度,摇匀,配制成浓度为152.40 μgmL[sup]-[/sup][sup]1[/sup]的DHEA标准品储备溶液。[b][b]2.2样品的处理[/b][/b]2.2.1 腺苷样品的处理取本品粉末(过3号筛)约1.0 g,精密称定,置具塞锥形瓶中,精密加入15%甲醇l0 mL,密塞,摇匀,称定质量,超声处理30 min,放冷,再称定质量,用15%甲醇补足减失的重量,12000 rmin[sup]-[/sup][sup]1[/sup]离心15 min,取上清液,0.45 μm微孔滤膜滤过,取续滤液作为供试品溶液。2.2.2 DHEA样品的处理取本品粉末(过3号筛)约3.0 g,精密称定,置具塞锥形瓶中,精密加入甲醇l5 mL,密塞,摇匀,称定质量,超声处理1 h,放冷,再称定质量,用甲醇补足减失的重量,摇匀,0.45μm微孔滤膜滤过,取续滤液作为供试品溶液。[b][b]2.3 色谱条件[/b][/b]2.3.1 腺苷色谱条件:色谱柱:Aglient Extend XDB C18柱(4.6 mm×250 mm,5 μm);流动相:磷酸盐缓冲液(pH6.5) -甲醇(94:6,[i]v/v[/i]);流速:1 mLmin[sup]-[/sup][sup]1[/sup];柱温:30 °C; 检测波长:260 nm;进样量:10 μL。HPLC图见图1。2.3.2 DHEA色谱条件:色谱柱:Welch Ultimate XB-C18柱(4.6 mm×250 mm,5 μm);流动相:甲醇-水(62:38,[i]v/v[/i]);流速:0.6 mLmin[sup]-[/sup][sup]1[/sup];柱温:30 °C; 检测波长:204 nm;进样量:10 μL。HPLC图见图2。[img=,690,278]https://ng1.17img.cn/bbsfiles/images/2019/07/201907031540228286_5423_3451954_3.jpg!w690x278.jpg[/img][img=,690,300]https://ng1.17img.cn/bbsfiles/images/2019/07/201907031540280876_3707_3451954_3.jpg!w690x300.jpg[/img][b][b]2.4标准曲线的绘制[/b][/b]精密吸取腺苷标准品储备溶液0.5,1,3,5,8,10 mL分别置于10 mL量瓶中,加甲醇定容至刻度,摇匀,按上述色谱条件进样,以峰面积值 Y为纵坐标,腺苷浓度X为横坐标绘制标准方程为:[i]Y[/i]=34197.2[i] X[/i]+25515.1,[i]r[/i]=0.9999,表明腺苷在13.22 ~ 264.40 μgmL[sup]-[/sup][sup]1[/sup]浓度内与峰面积线性关系良好。精密吸取DHEA标准品储备溶液1 mL置于10 mL量瓶中,加甲醇定容至刻度,摇匀,得15.24 μgmL[sup]-[/sup][sup]1[/sup]的DHEA使用溶液。分别精密吸取DHEA使用溶液0.5,1,3,5,8,10 mL置于10 mL量瓶中,加甲醇定容至刻度,摇匀,按上述色谱条件进样,以峰面积值 Y为纵坐标,DHEA浓度X为横坐标绘制标准方程为:[i]Y[/i]=13042.7 [i]X[/i]-1836.3,[i]r[/i]=0.9999,表明DHEA在0.76 ~ 15.24 μgmL[sup]-[/sup][sup]1[/sup]浓度内与峰面积线性关系良好。[b][b]2.5 重复性试验[/b][/b] 取同一供试品粉末6份,精密称定,按“2.2”项下方法处理,按“2.3”项下色谱条件测定峰面积。结果腺苷和DHEA峰面积的RSD分别为1.87 %和1.91 % (n=6),表明方法重复性良好。[b][b]2.6 精密度试验[/b][/b]精密吸取同一供试品溶液10 μL,按上述色谱条件连续进样6次,测定腺苷、DHEA峰面积值,其RSD分别为0.92 %和1.03 % (n=6),表明仪器及进样精密度良好。[b][b]2.7 稳定性试验[/b][/b] 精密吸取同一供试品溶液,室温放置,分别在0,2,4,8,12,24,48 h进样分析。测定腺苷、DHEA峰面积值,其RSD分别为2.07 %和1.65 % (n=6),表明供试品溶液在48 h内稳定性良好。[b][b]2.8 回收率[/b][/b]分别称取已知含量的样品6份,精密称定,分别加入相当含量的腺苷和DHEA对照品溶液,按“2.2”项下供试品溶液的制备方法制备供试品溶液,分别进行测定,计算回收率。结果腺苷、DHEA平均加样回收率为98.10 % (RSD=1.40 %)和97.72 % (RSD=1.15 %),结果见表1、2。[align=center][b]表1 山药样品中腺苷的加样回收率实验结果(n=6)[/b][/align][table][tr][td][align=center]取样量/g[/align][/td][td][align=center]样品中量/mg[/align][/td][td][align=center]添加量/mg[/align][/td][td][align=center]测得量/mg[/align][/td][td][align=center]回收率/%[/align][/td][td][align=center]平均回收率/%[/align][/td][td][align=center]RSD/%[/align][/td][/tr][tr][td][align=center]0.5014[/align][/td][td][align=center]0.1604 [/align][/td][td][align=center]0.1586[/align][/td][td][align=center]0.3141[/align][/td][td][align=center]96.88 [/align][/td][td=1,6][align=center]98.10[/align][/td][td=1,6][align=center]1.40[/align][/td][/tr][tr][td][align=center]0.5011[/align][/td][td][align=center]0.1604 [/align][/td][td][align=center]0.1586[/align][/td][td][align=center]0.3201[/align][/td][td][align=center]100.72 [/align][/td][/tr][tr][td][align=center]0.4989[/align][/td][td][align=center]0.1596 [/align][/td][td][align=center]0.1586[/align][/td][td][align=center]0.3147[/align][/td][td][align=center]97.76 [/align][/td][/tr][tr][td][align=center]0.4997[/align][/td][td][align=center]0.1599 [/align][/td][td][align=center]0.1586[/align][/td][td][align=center]0.3141[/align][/td][td][align=center]97.22 [/align][/td][/tr][tr][td][align=center]0.5001[/align][/td][td][align=center]0.1600 [/align][/td][td][align=center]0.1586[/align][/td][td][align=center]0.3159[/align][/td][td][align=center]98.28 [/align][/td][/tr][tr][td][align=center]0.5012[/align][/td][td][align=center]0.1604 [/align][/td][td][align=center]0.1586[/align][/td][td][align=center]0.3154[/align][/td][td][align=center]97.74 [/align][/td][/tr][/table][align=center][b]表2 山药样品中DHEA的加样回收率实验结果(n=6)[/b][/align][table][tr][td][align=center]取样量/g[/align][/td][td][align=center]样品中量/mg[/align][/td][td][align=center]添加量/mg[/align][/td][td][align=center]测得量/mg[/align][/td][td][align=center]回收率/%[/align][/td][td][align=center]平均回收率/%[/align][/td][td][align=center]RSD/%[/align][/td][/tr][tr][td][align=center]1.5013[/align][/td][td][align=center]0.0293 [/align][/td][td][align=center]0.0305[/align][/td][td][align=center]0.0589[/align][/td][td][align=center]97.13 [/align][/td][td=1,6][align=center]97.72 [/align][/td][td=1,6][align=center]1.15 [/align][/td][/tr][tr][td][align=center]1.5024[/align][/td][td][align=center]0.0293 [/align][/td][td][align=center]0.0305[/align][/td][td][align=center]0.0591[/align][/td][td][align=center]97.72 [/align][/td][/tr][tr][td][align=center]1.4997[/align][/td][td][align=center]0.0292 [/align][/td][td][align=center]0.0305[/align][/td][td][align=center]0.0597[/align][/td][td][align=center]99.86 [/align][/td][/tr][tr][td][align=center]1.5001[/align][/td][td][align=center]0.0293 [/align][/td][td][align=center]0.0305[/align][/td][td][align=center]0.0589[/align][/td][td][align=center]97.21 [/align][/td][/tr][tr][td][align=center]1.4987[/align][/td][td][align=center]0.0292 [/align][/td][td][align=center]0.0305[/align][/td][td][align=center]0.0587[/align][/td][td][align=center]96.64 [/align][/td][/tr][tr][td][align=center]1.5011[/align][/td][td][align=center]0.0293 [/align][/td][td][align=center]0.0305[/align][/td][td][align=center]0.0591[/align][/td][td][align=center]97.80 [/align][/td][/tr][/table][b][b]2.9 结果分析[/b][/b]精密称取不同来源的山药粉末,按“2.2”项下方法制备供试品溶液,分别进样,记录峰面积积分值,外标法计算腺苷及DHEA的含量。结果如下表3所示。[align=center][b]表3 不同来源山药药材中腺苷及DHEA的含量([i]n[/i]=3)[/b][/align][table][tr][td][align=center]编号[/align][/td][td][align=center]样品来源[/align][/td][td][align=center]腺苷含量/%[/align][/td][td][align=center]DHEA含量/mg/kg[/align][/td][/tr][tr][td][align=center]1[/align][/td][td][align=center]种植铁棍山药[/align][/td][td][align=center]0.032[/align][/td][td][align=center]19.50 [/align][/td][/tr][tr][td][align=center]2[/align][/td][td][align=center]野生山药1[/align][/td][td][align=center]0.027[/align][/td][td][align=center]5.38 [/align][/td][/tr][tr][td][align=center]3[/align][/td][td][align=center]野生山药2[/align][/td][td][align=center]0.024[/align][/td][td][align=center]3.29[/align][/td][/tr][tr][td][align=center]4[/align][/td][td][align=center]驯化野生山药雌株[/align][/td][td][align=center]0.037[/align][/td][td][align=center]24.05 [/align][/td][/tr][tr][td][align=center]5[/align][/td][td][align=center]驯化野生山药雄株[/align][/td][td][align=center]0.040[/align][/td][td][align=center]62.25 [/align][/td][/tr][tr][td][align=center]6[/align][/td][td][align=center]紫山药[/align][/td][td][align=center]ND[/align][/td][td][align=center]ND[/align][/td][/tr][/table] 从测定结果看,不同来源山药中腺苷和DHEA含量差别较大,可能由于品种及生长环境的差异造成。研究表明,驯化野生山药雄株中腺苷和DHEA含量最高,显著高于野生山药中的含量,说明驯化过程对野生山药的改良起到了很好的积极作用。另外,驯化野生山药雄株中DHEA含量比公认的营养成分较高的铁棍山药高出3倍左右,进一步说明该山药品质的优异性。 另外,紫山药中并未检测到腺苷和DHEA两种成分的存在,这可能由于紫山药自身品种差异有关。[b][b]3 小结和讨论[/b][/b]3.1 DHEA提取条件优化 在DHEA测定前处理时,本实验曾根据李军等人的优化方法,采用甲醇-丙酮(6:4)超声提取、水浴蒸干、二氯甲烷多次萃取等主要工序对山药中DHEA进行提取,该方法提取效果较好,DHEA可以被较好的富集和检测。但是由于提取过程复杂,实验重复性和加样回收率较难控制。本实验采用甲醇、乙醇、甲醇-丙酮(1:1,[i]v/v[/i])等溶剂对山药样品进行超声1h提取,结果发现,四种溶剂提取效果没有明显差异,因此选用甲醇做为最终提取溶剂。 本实验采用的方法大大简化了山药中DHEA的提取过程,减少了有毒试剂的使用,缩短了提取时间,且提取率与李军的方法相比没有显著的差异。3.2 小结 本研究对山药中腺苷和DHEA成分的提取和测定条件进行优化,为山药质量标准建立和质量评价提供参考依据。另外,研究对包括野生、种植、驯化品种等不同来源山药中腺苷和DHEA指标含量进行测定,为山药品种的选育和驯化提供一定的研究基础。
丙酮氰醇,又名2-羟基-2-甲基丙腈,是一种有机化合物,化学式为C4H7NO,有剧毒 [1] ,用于制甲基丙烯酸甲酯、偶氮二异丁腈等,还用作合成农药。 理化性质 密度:0.932g/cm3 熔点:-19℃ 沸点:120℃(分解) 闪点:63℃ 蒸汽压:3.07kPa(82℃) 外观:无色至淡黄色液体 溶解性:能与水、醇、醚及其他多种有机溶剂混溶,不溶于石油醚和二硫化碳
如题,检测化合物为多溴联苯醚,标品溶剂为异辛烷,前处理要求必须用乙醇或丙酮,不能用异辛烷或正己烷。就怕影响定性或是定量啊!求大家帮助!
如题,检测化合物为多溴联苯醚,标品溶剂为异辛烷,前处理要求必须用乙醇或丙酮,不能用异辛烷或正己烷。就怕影响定性或是定量啊!求大家帮助!
我做生物胺的柱前衍生,用的是10mg/ml的丹磺酰氯丙酮容液,但是配完之后丹磺酰氯很多都沉在底部并不溶解,用超声混匀后,用移液枪吸取能明显看到颗粒。然后将丹磺酰氯丙酮容液加进生物胺单标里混匀后会底部出现白色沉淀,是不是有发生什么化学反应?这个沉淀看了很多文献都没提到过。然后就是去跑液相,8个标品,只有精氨酸一直跑不出,时间感觉也很充足了,就是一直很平坦不出峰,是不是和上面的操作有关系啊?
[b]《空气和废气监测分析方法》(第四版增补版) 环氧氯丙烷 乙酰丙酮分光光度法 [b]环氧氯丙烷的标准曲线哪个大神有?[/b]扩项急用,万分感谢[/b]
用溶剂解吸法测定工作场所空气中的苯、丙酮等有害物质时,用到的苯、丙酮等GCS级的标准物质都是低沸点,挥发性强的化合物,一般密封在安瓿瓶中,取用后,不知如何保存?我用过一瓶2mL的苯,敲开安瓿瓶,取完样后,用棉花堵住口子,再包上生料带,放在0~5℃的冰箱中保存,可过了约半个月,发现安瓿瓶中的苯全部挥发完了,不知大家如何保存?
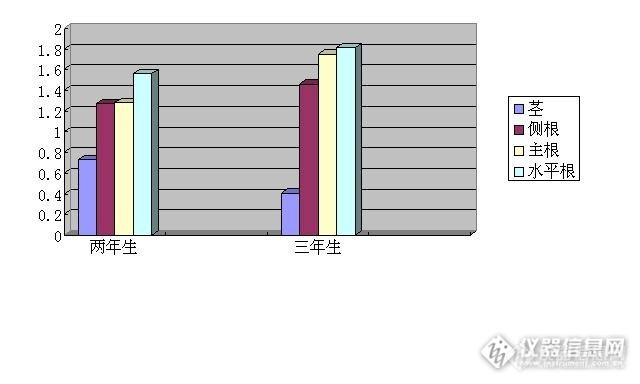
本实验分别提取两年生、三年生无腺毛甘草主根、侧根、水平根及茎中的黄酮,并测定其含量,初步找出消长规律。以甲醇为提取溶剂,索氏提取法提取黄酮,分光光度法测定总黄酮含量。结果表明两年生无腺毛中主根、侧根、水平根及茎中的黄酮含量分别为1.28%、1.28%、1.56%、0.73%;三年生无腺毛中主根、侧根、水平根及茎中的黄酮含量分别为1.75%、1.46%、1.81%、0.41%。随着年限的增大,无腺毛甘草中根部的黄酮含量逐渐增大。同龄期无腺毛甘草中黄酮含量:水平根最高,主根和侧根次之,茎最小。 无腺毛甘草 黄酮 含量测定 消长规律1.前 言无腺毛甘草(Glycyrrhiza eglandulosa X.Y.Li)是石河子大学李学禹教授在新疆发现、1993年命名的新种,也是我国的特有种。它生长的环境条件比乌拉尔甘草(G. uralensis)更恶劣,因而更加耐干旱、耐盐碱,而且是很适合于低产地、弃耕地等恶劣环境条件下生长的抗逆性极强的甘草物种。这个种在形态分类系统学、细胞学、生态学都有研究,根据细胞染色体组型分析与形态数值分析,都证实它与乌拉尔甘草、光果甘草亲缘关系较近,但在化学成分方面未进行系统研究。根据资料报导,产于新疆的胀果甘草(G.inflata)、黄甘草(G.eurycarpa)及云南的云南甘草(G.yunnanensis)都发现过大量的具有生理活性的新的化学成分。由此可知:一个形态性状特异、分布于逆境条件下能正常生长的新物种,肯定具有抗逆性较强的基因所决定的代谢产物,肯定有特殊性。为此,我们拟从应用开发的角度,提取无腺毛甘草中黄酮类化合物,对其进行含量测定,并进一步研究其在不同龄期、不同部位黄酮类化合物的消长规律,为进一步研究其化学组成和结构打下基础,并且为退耕还草大面积栽培无腺毛甘草提供科学依据。2.实验部分2.1实验仪器、样品与试剂2.1.1实验仪器:紫外可见分光光度计(上海棱光技术有限公司),索氏提取器(自治区化玻站提供),烘干箱(湖北省黄石医疗器材厂),电光分析天平(上海棱光技术有限公司),精密PH计(上海雷磁仪器厂)。2.1.2实验样品:两年生、三年生的无腺毛甘草(石河子大学甘草栽培基地李学禹教授提供)。2.1.3实验试剂:柚皮苷对照品(中国药品生物制品检定所),10%的氢氧化钾溶液,甲醇(AR)。2.2黄酮类化合物的提取分别称取3份两年生无腺毛甘草的主根粉碎后放入索氏提取器中,加入甲醇回流2h,冷却后分别将回流液转移至容量瓶中,提取两次,提取液合并,用甲醇定容。用同样的方法分别提取两年生无腺毛甘草的水平根、侧根和茎以及三年生无腺毛甘草中主根、侧根、水平根、茎中的黄酮。2.3黄酮类化合物的含量测定2.3.1对照品溶液的制备准确称取柚皮苷对照品适量,用甲醇溶解并定容于10mL容量瓶中,制得浓度约为1.0mg.mL-1的对照品溶液。2.3.2最大吸收峰的确定精确吸取柚皮苷对照品溶液0.5ml,加入5ml甲醇,再加入10%KOH溶液2.5ml,室温放置5min,用甲醇稀释至50ml[
请问芳香族羧酸类化合物GCMS能定性么?今天收到一个粉末样品,可能是芳香族羧酸类化合物,甲醇/丙酮/正己烷/氯仿/甲苯都尝试了一遍,只有甲醇溶解较好,但是仍不能完全溶解,取甲醇溶液过滤后走了GC-FID,并无明显峰检出,客户要求GCMS定性,这样怎么走MS呢?请大家帮帮忙~谢谢
做一氯丙酮的时候,使用方法90度恒温,一氯丙酮出峰时间在2.0左右,在2.9分有一个峰怀疑是一氯丙酮缩合物,网上查不出资料,有没有知道2.9分那个峰的沸点是多少,最好有具体详细的物性介绍
“短而悍”[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=26334]有机化合物签别[/url]在药品的生产、研究及检验等过程中,常常会遇到有机化合物的分离、提纯和鉴别等问题。有机化合物的鉴别、分离和提纯是三个既有关联而又不相同的概念。 分离和提纯的目的都是由混合物得到纯净物,但要求不同,处理方法也不同。分离是将混合物中的各个组分一一分开。在分离过程中常常将混合物中的某一组分通过化学反应转变成新的化合物,分离后还要将其还原为原来的化合物。提纯有两种情况,一是设法将杂质转化为所需的化合物,另一种情况是把杂质通过适当的化学反应转变为另外一种化合物将其分离(分离后的化合物不必再还原)。 鉴别是根据化合物的不同性质来确定其含有什么官能团,是哪种化合物。如鉴别一组化合物,就是分别确定各是哪种化合物即可。在做鉴别题时要注意,并不是化合物的所有化学性质都可以用于鉴别,必须具备一定的条件:(1) 化学反应中有颜色变化(2) 化学反应过程中伴随着明显的温度变化(放热或吸热)(3) 反应产物有气体产生(4) 反应产物有沉淀生成或反应过程中沉淀溶解、产物分层等。 本课程要求掌握的重点是化合物的鉴别,为了帮助大家学习和记忆,将各类有机化合物的鉴别方法进行归纳总结,并对典型例题进行解析。 一.各类化合物的鉴别方法 1.烯烃、二烯、炔烃: (1)溴的四氯化碳溶液,红色腿去 (2)高锰酸钾溶液,紫色腿去。 2.含有炔氢的炔烃: (1) 硝酸银,生成炔化银白色沉淀 (2) 氯化亚铜的氨溶液,生成炔化亚铜红色沉淀。 3.小环烃:三、四元脂环烃可使溴的四氯化碳溶液腿色 4.卤代烃:硝酸银的醇溶液,生成卤化银沉淀;不同结构的卤代烃生成沉淀的速度不同,叔卤代烃和烯丙式卤代烃最快,仲卤代烃次之,伯卤代烃需加热才出现沉淀。 5.醇: (1) 与金属钠反应放出氢气(鉴别6个碳原子以下的醇); (2) 用卢卡斯试剂鉴别伯、仲、叔醇,叔醇立刻变浑浊,仲醇放置后变浑浊,伯醇放置后也无变化。 6.酚或烯醇类化合物: (1) 用三氯化铁溶液产生颜色(苯酚产生兰紫色)。 (2) 苯酚与溴水生成三溴苯酚白色沉淀。 7.羰基化合物: (1) 鉴别所有的醛酮:2,4-二硝基苯肼,产生黄色或橙红色沉淀; (2) 区别醛与酮用托伦试剂,醛能生成银镜,而酮不能; (3) 区别芳香醛与脂肪醛或酮与脂肪醛,用斐林试剂,脂肪醛生成砖红色沉淀,而酮和芳香醛不能; (4) 鉴别甲基酮和具有结构的醇,用碘的氢氧化钠溶液,生成黄色的碘仿沉淀。 8.甲酸:用托伦试剂,甲酸能生成银镜,而其他酸不能。 9.胺:区别伯、仲、叔胺有两种方法 (1)用苯磺酰氯或对甲苯磺酰氯,在NaOH溶液中反应,伯胺生成的产物溶于NaOH;仲胺生成的产物不溶于NaOH溶液;叔胺不发生反应。 (2)用NaNO2+HCl: 脂肪胺:伯胺放出氮气,仲胺生成黄色油状物,叔胺不反应。 芳香胺:伯胺生成重氮盐,仲胺生成黄色油状物,叔胺生成绿色固体。 10.糖: (1) 单糖都能与托伦试剂和斐林试剂作用,产生银镜或砖红色沉淀; (2) 葡萄糖与果糖:用溴水可区别葡萄糖与果糖,葡萄糖能使溴水褪色,而果糖不能。 (3)麦芽糖与蔗糖:用托伦试剂或斐林试剂,麦芽糖可生成银镜或砖红色沉淀,而蔗糖不能。 二.例题解析 例1.用化学方法鉴别丁烷、1-丁炔、2-丁炔。 分析:上面三种化合物中,丁烷为饱和烃,1-丁炔和2-丁炔为不饱和烃,用溴的四氯化碳溶液或高锰酸钾溶液可区别饱和烃和不饱和烃,1-丁炔具有炔氢而2-丁炔没有,可用硝酸银或氯化亚铜的氨溶液鉴别。因此,上面一组化合物的鉴别方法为: 例2.用化学方法鉴别氯苄、1-氯丙烷和2-氯丙烷。 分析:上面三种化合物都是卤代烃,是同一类化合物,都能与硝酸银的醇溶液反应生成卤化银沉淀,但由于三种化合物的结构不同,分别为苄基、二级、一级卤代烃,它们在反应中的活性不同,因此,可根据其反应速度进行鉴别。上面一组化合物的鉴别方法为: 例3.用化学方法鉴别下列化合物 苯甲醛、丙醛、2-戊酮、3-戊酮、正丙醇、异丙醇、苯酚 分析:上面一组化合物中有醛、酮、醇、酚四类,醛和酮都是羰基化合物,因此,首先用鉴别羰基化合物的试剂将醛酮与醇酚区别,然后用托伦试剂区别醛与酮,用斐林试剂区别芳香醛与脂肪醛,用碘仿反应鉴别甲基酮;用三氯化铁的颜色反应区别酚与醇,用碘仿反应鉴别可氧化成甲基酮的醇。鉴别方法可按下列步骤进行: (1) 将化合物各取少量分别放在7支试管中,各加入几滴2,4-二硝基苯肼试剂,有黄色沉淀生成的为羰基化合物,即苯甲醛、丙醛、2-戊酮、3-戊酮,无沉淀生成的是醇与酚。 (2) 将4种羰基化合物各取少量分别放在4支试管中,各加入托伦试剂(氢氧化银的氨溶液),在水浴上加热,有银镜生成的为醛,即苯甲醛和丙醛,无银镜生成的是2-戊酮和3-戊酮。 (3) 将2种醛各取少量分别放在2支试管中,各加入斐林试剂(酒石酸钾钠、硫酸酮、氢氧化钠的混合液),有红色沉淀生成的为丙醛,无沉淀生成的是苯甲醛。 (4) 将2种酮各取少量分别放在2支试管中,各加入碘的氢氧化钠溶液,有黄色沉淀生成的为2-戊酮,无黄色沉淀生成的是3-戊酮。 (5) 将3种醇和酚各取少量分别放在3支试管中,各加入几滴三氯化铁溶液,出现兰紫色的为苯酚,无兰紫色的是醇。 (6) 将2种醇各取少量分别放在支试管中,各加入几滴碘的氢氧化钠溶液,有黄色沉淀生成的为异丙醇,无黄色沉淀生成的是丙醇。
做蔬菜样品时,先加的水和丙酮,加NaCl分层后,上层应该是丙酮吧?但为什么开始加的丙酮比水多很多,分层后丙酮却比水相少很多呢?而且色素全在丙酮里。 分出来的水相再加二氯甲烷提,分层后下层是二氯甲烷吗?怎么也有人说加了NaCl使二氯甲烷在水的上层呢?

10,抽取5个版友);中奖名单:999youran(注册ID:999youran)zgx3025(注册ID:v2844608)m3071659(注册ID:m3071659)馨语(注册ID:huangdm)莫名其妙(注册ID:moyueqiu)http://ng1.17img.cn/bbsfiles/images/2016/09/201609261647_612177_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609261647_612178_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================三磷酸腺苷二钠注射液方法:HPLC应用编号:101389基质:药品应用编号:101389化合物:三磷酸腺苷二钠固定相:Platisil ODS色谱柱/前处理小柱:Platisil ODS 5u 150 x 4.6 mm样品前处理:【含量测定】三磷酸腺苷二钠的重量比 取本品适量,精密称定,加流动相溶解并定量稀释成每1ml中含0.4mg的溶液。色谱条件:检测波长:UV 259 nm 流动相:0.2mol/L磷酸盐缓冲溶液-甲醇(95:5) 缓冲溶液:取磷酸氢二钠35.8g、磷酸二氢钾13.6g,加水900ml溶解,用1mol/L氢氧化钠溶液调pH至7.0,加入四丁基溴化铵1.61g,加水至1000ml,摇匀。 洗脱方式:等度 柱温:35℃ 进样量:10 ul文章出处:P29关键字:三磷酸腺苷二钠注射液,2010版中国药典,HPLC,含量测定,铂金,Platisil ODS谱图:http://www.dikma.com.cn/Public/Uploads/images/atp(1).PNG图例:1. 一磷酸腺苷二钠;2. 二磷酸腺苷二钠;3. 三磷酸腺苷二钠
如题,公司接了一个项目,要测空气中的丙酮,根据HJ683测定丙烯醛和丙酮峰无法分离,这种情况,HJ683是不是就不适用?还是说可以用,就出丙烯醛和丙酮的总量?请各位大佬指点,谢谢了[img=,690,407]https://ng1.17img.cn/bbsfiles/images/2021/09/202109071703185804_8826_4026365_3.png!w690x407.jpg[/img][img=,690,516]https://ng1.17img.cn/bbsfiles/images/2021/09/202109071703222548_8658_4026365_3.png!w690x516.jpg[/img]
三氯杀螨醇在丙酮中的稳定性 关于在有机溶剂中农药稳定性问题,有人发现:三氯杀螨醇标液在棕色小瓶室温下持续6个小时的稳定性试验,三氯杀螨醇在丙酮中迅速降解。大家平时实验时有没有发现这种现象?
参评论文题目:全自动阵列离子迁移谱仪连续监测挥发性有机化合物。论文概述: 为了拓宽离子迁移谱仪的检测范围、提高化合物的识别准确度,研制了一套阵列离子迁移谱仪,该仪器基于63Ni源正离子模式、63Ni源负离子模式和真空紫外灯光电离模式的组合电离源,可以连续监测空气中挥发性有机化合物。仪器采用全自动的采样进样系统,同时检测了二甲基亚砜的正离子和二氯甲烷的负离子,实现了正负离子的同时检测。通过对阵列离子迁移谱图的综合解析,识别了63Ni源正离子模式下难以鉴别的丙烯腈、间二甲苯和丙酮。连续4 d定量测定丙酮样品,结果表明仪器对丙酮的线性检测范围为2个数量级,线性相关系数R优于0.995,相对标准偏差控制在4.0%~18.3%。采用动态跟踪法,连续24 h在线监测了模拟泄漏的丙烯酸甲酯,监测结果直接反映了其泄漏的时间和浓度。







 我要推广仪器
我要推广仪器
 下载APP
下载APP