[color=#444444]环己烯水合制备环己醇的实验,反应结束后用乙酸乙酯萃取,最终得到含有环己烯、环己醇、乙酸乙酯的有机相(应该有一些副产物)。[/color][color=#444444] [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析产物中环己烯和环己醇的质量,来确定环己烯的转化率和反应选择性。准备用内标法,没确定好用什么内标物,最好是毒性比较小的,望大神相助!谢谢![/color]
以下所述校正因子均为FID中物质相对正庚烷的相对校正因子! 查资料环己酮的FID校正因子为1.38,但是环己醇仅有TCD的校正因子,无FID的校正因子。 若按照有效碳数法计算,查资料羰基贡献为0,即环己酮校正因子按有效碳数法计算:7*98/(100*5)=1.37。请问下羟基的贡献是多少呢,环己醇FID校正因子是多少? 文献理论上看,羰基贡献为0,羟基贡献应大于零,也就是说环己醇的响应因子应比环己酮大,也就是环己醇的校正因子比环己酮小。 仪器实测环己酮校正因子1.20,环己醇1.11。 求帮助!
7月2日将50ug/ml的环己基氨基磺酸钠标准溶液在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]上机测试,得出色谱图。 然后将50ug/ml的环己基氨基磺酸钠标准溶液分为两部分,分别储存在冷冻和冷藏的条件下密封保存。 在7月10日的时候,分别取出在[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]上机测试,得出色谱图。 将三个环己基氨基磺酸钠的色谱图进行对比。 [img=,690,309]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161522359457_3024_5979722_3.png!w690x309.jpg[/img] 环己基氨基磺酸钠的色谱图会出现两个色谱峰,分别是环己醇亚硝酸酯、环己醇。 我发现两个问题 1、7月10日的色谱图和7月2日的色谱图相比(期间其他条件都没有变化),环己醇亚硝酸酯、环己醇的出峰位置都提前了5-10秒,算是色谱峰漂移了。理论上应该是再跑一个当天衍生化的环己基氨基磺酸钠标准溶液上机进行对比,但是近期比较忙这一本就没有做。后期有机会做了,我再反馈这一个问题。 2、在冷藏和冷冻条件下的环己基氨基磺酸钠标准溶液的环己醇亚硝酸酯、环己醇的面积有一些差别。冷藏条件和冷冻条件相比,冷藏条件下的环己醇亚硝酸酯的面积变小了,环己醇的面积变大了。但是面积总和相比几乎一样,因此可以判定,环己醇亚硝酸酯在指定温度下,是可以继续转化为环己醇的。 [img=,690,511]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161523569214_3530_5979722_3.png!w690x511.jpg[/img] [img=,690,513]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161524065331_7755_5979722_3.png!w690x513.jpg[/img] [img=,690,535]https://ng1.17img.cn/bbsfiles/images/2024/08/202408161524102083_9380_5979722_3.png!w690x535.jpg[/img] 根据标准曲线查看7月10日分别上机的环己基氨基磺酸钠标准溶液的含量,分别是58.7689 ug/ml和58.5511 ug/ml,7月2日上机检测的含量是51.2462 ug/ml,我们可以看出来标准溶液保存8天后,检测的数值就有不少误差了,而且是数值变大了。这一点不知道是什么原因
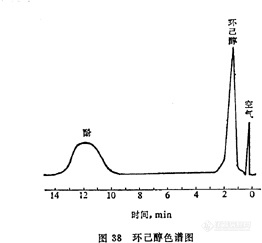
空气中环己醇的测定方法 1 原理空气中环己醇、酚经己二酸乙二醇聚酯柱分离后,用氢焰离子化检测器检测,以保留时间定性,峰高定量。2 仪器2.1 注射器,100ml,1ml。2.2 微量注射器,1微升。2.3 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],氢焰离子化检测器。5ng环己醇给出的信噪比不低于3∶1。色谱柱:柱长2m,内径4mm不锈钢柱。己二酸乙二醇聚酯:201担体=15∶100柱温:120℃汽化室温度:170℃检测室温度:140℃载气(氮气):48ml/min3 试剂3.1 环己醇,色谱纯。3.2 己二酸乙二醇聚酯,色谱固定液。3.3 201担体,40~60目。4 采样取100ml注射器,在采样点用现场空气抽洗3次,然后抽取100ml空气,将注射器套上橡皮帽,垂直放置,当天分析。5 分析步骤5.1 对照试验:将100ml注射器取下塑料帽,抽取100ml清洁空气,与样品同时分析,作为对照。5.2 样品处理:将样品与对照样品注射器垂直放置,记录实验室的温度和气压。5.3 标准曲线的绘制:用微量注射器准确抽取一定量的环己醇(于20℃时1微升环己醇质量为0.9624mg)注入100ml注射器中,配成一定浓度的标准气体。取一定量的上述标准气体用空气稀释成0.025、0.050、0.1、0.5微克/ml的标准气体。[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201514_52393_1625938_3.jpg[/img]分别取1ml进样,测量保留时间及峰高,每个浓度重复3次,取峰高的平均值,以环己醇的含量对峰高作图,绘制标准曲线。保留时间为定性指标。5.4 测定:取1ml空气样品直接进样,用保留时间定性,峰高定量。6 计算X=C*1000/V0式中:X——空气中环己醇的浓度,mg/m3;C——由标准曲线上查出的环己醇含量,微克;V0——标准状况下的样品体积,ml。7 说明7.1 本法不能将环己醇与环己酮分离。7.2 本法检测限为5ng。
求助:工业级环己醇测定的相关国标
[color=#444444]双氧水做氧化剂,杂多酸做催化剂氧化环己醇,反应产物主要是环己酮([url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析)外,还有己二酸,除此之外还有什么产物?越具体越好,分别用用什么方法定性定量分析?[/color]
先说结论:衍生化后的环己烷氨基磺酸钠标准溶液在25摄氏度下5个小时就会有变化,主色谱峰[font=&]环己醇的面积会变小,衍生物色谱峰环己醇亚硝酸酯的面积会变大(应该是两者面积和不会变化,理论上还是可以继续用来做标准曲线的)。而且低浓度的环己烷氨基磺酸钠标准溶液衍生化后的色谱峰环己醇的色谱峰会出现杂峰。 我猜测高浓度的环己烷氨基磺酸钠标准溶液衍生化后的色谱峰环己醇的色谱峰不会出现杂峰是因为主峰比较大,把杂峰包裹进去了。 [/font] 上午对环己烷氨基磺酸钠标准溶液进行衍生化处理,浓度分别是10μg/ml、50μg/ml、500μg/ml,处理完毕后立刻上机。一共是12个进样瓶,第一次上机最后一个样品跑完后,大概时间是中午12:30,然后等待下午2:30的时候,又重新上机。 每个进样瓶的信号采集时间大概是20分钟,这样第一个样品和最后一个样品间隔时间是20*11=220分钟,加上中午的2个小时,这样大概是5个小时。也就是说第一批和第二批进样中每个相同序号的进样瓶采集时间间隔是5小时 这次使用的环己烷氨基磺酸钠标准溶液有两种,分别是10个月前配置的标准溶液和新配置的标准溶液。 分别编号是10-1、50-1、500-1、10-2、50-2、500-2 下面是各个对比的色谱图,分别是上午的色谱图和下午的色谱图进行对比 10-1的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇旁边出现了杂峰 [img=,690,373]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181124064647_4366_5979722_3.png!w690x373.jpg[/img] 50-1的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇面积明显变小了,衍生物色谱峰环己醇亚硝酸酯的面积明显变大了。 [img=,690,457]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181124061287_876_5979722_3.png!w690x457.jpg[/img] 500-1的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,500μg/ml的这个浓度上午和下午的色谱图区别不大 [img=,690,446]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181124064695_5130_5979722_3.png!w690x446.jpg[/img] 10-2的对比,上午和下午的色谱图进行对比,环己烷氨基磺酸钠标准溶液的主色谱峰旁边出现了杂峰 [img=,690,413]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181150579387_7325_5979722_3.png!w690x413.jpg[/img] 50-2的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇面积明显变小了,衍生物色谱峰环己醇亚硝酸酯的面积明显变大了。 [img=,690,481]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181150587943_3504_5979722_3.png!w690x481.jpg[/img] 500-2的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇面积明显变小了,衍生物色谱峰环己醇亚硝酸酯的面积明显变大了。 [img=,690,509]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181150579543_2309_5979722_3.png!w690x509.jpg[/img]
请教各位谁知道哪里能买到棉酚标准品和3-氨基-1-丙醇?谢谢!急用
老师您好!关于环己烷氧化产物的分析我还有一个问题要请教:环己烷氧化的主要产物是环己醇、环己酮、己二酸以及己二酸二环己醇酯。我目前需要测定这些氧化产物的浓度,以确定氧化过程动力学方程。关于前两种产物,文献中已有现成的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析法;关于后两种产物酸、酯的分析,却依然在采用误差比较大、费时比较久的化学滴定法。我想放弃化学分析法,而采用可能误差会小一些而且又切实可行的某种仪器分析法比如高效液相色谱,但不知是否可行而且也不知道该如何去寻找合适的色谱条件和具体分析方法。请前辈多多指教!
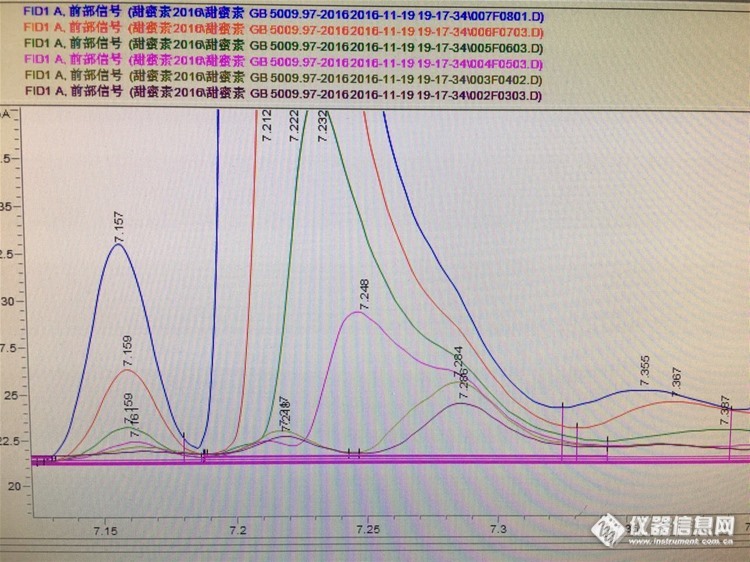
请问有人做GB 5009.97-2016的甜蜜时光素吗?为什么浓度低的环己醇的峰很奇怪……好友回复:进的标品的话,那就只能舍弃这个点了,这不关浓度的事,因为各物质都存在响应的高低,既然标品都成这样了,如果配置过程中没问题,那就是这个浓度不行了,你先再老化一下柱子吧,你重新割一下柱子,并且确保切口平整,柱子伸进进样口最好5mm这样,先从高浓度开始进样,试试看吧大家说说看~http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669708_3150883_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016112217295387_01_3150883_3.jpg
我要分离苯 环己烷 环己醇 环己烯 采用GC1690 SE-54毛细管色谱柱 分离条件如何选择啊
要测定环己醇和环己酮的校正因子?该用什么做内标物比较好?对柱温等其他温度该怎样设定比较好?另外麻烦高手教一下怎样设置程序升温啊
分离苯 环己烷 环己醇 环己烯 采用GC1690 SE-54毛细管色谱柱 分离条件如何选择啊
我的原料是辛烯、产物为1,2-辛二醇。内标想用环己醇。[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]是北分的SP-3420A。我用什么色谱柱合适?
各位专家,谁能赐教分析下列溶液组分中的苯胺和DAM含量可以采用什么分析方法?溶液组分:氯化钠、氢氧化钠、苯胺、DAM、环己胺、环己醇
在前面的问题中,混合溶液中的苯胺、DAM、含量在5ppm左右。环己胺、环己醇的含量在2ppm以下
查资料,只搞清楚了用内标曲线法进行半定量。我是用SPME测果实香气,选了环己醇或者3-辛醇做内标。 内标跟标准品的溶剂要尽量一致对吧?那用什么来做溶剂溶解内标呢? SPME测果实香气是气体进样,做内标曲线的时候,配制不同梯度的标准品加上内标是否也是气体进样? 麻烦大神回答一下,万分感谢!自己摸索点东西太不容易了! 另外,如何通过特征离子峰,确定图谱中某化合物的浓度?
SPME测定果实香气,进行半定量,选了环己醇或者3-辛醇做内标,内标跟标准品的溶剂要尽量一致对吧?那用什么溶剂来溶解内标呢?正己烷?SPME测定果实香气是气体进样,做内标曲线的时候,配制不同梯度的标准品再加内标,这一步的话是液体进样还是气体进样?另外,如何根据特征离子峰来确定该化合物的相对浓度?望大神不吝赐教!感谢!!!

食品中甜蜜素测定心得食品中甜蜜素测定方法一般都是按照GB/T5009.97-2003进行的,方法的原理是在硫酸介质中环己基氨基磺酸钠与亚硝酸钠反应,生成环己醇亚硝酸酯,利用气相色谱法进行定量测定。而在实际工作中发现,环己醇亚硝酸酯主峰并不稳定,每次做标准曲线线性都不是很理想,于是考察了一下其稳定性。连续进同一个浓度的标准品,发现主峰的面积在不断减小,后面随之出现了一个小峰,并且在逐渐增大。通过用质谱进行定性发现,小峰是环己醇。通过资料查询得知,环己醇亚硝酸酯的确不稳定,会在衍生后逐渐转变成环己醇。这就是为什么同时出现两个峰,主峰不断减小,后面的小峰逐渐变大的缘故。当发现这个问题之后,似乎还是没有办法从根本上解决这个问题。因为在平时的工作中,一般都是批量处理样品和标准品。现在气相色谱都是自动序列进样,只要处理好样品之后放在样品盘上就可以依次自动进样测定了。这个就出现了一个问题。在这个过程中,不管是样品还是标准品中,都存在这一个潜移默化的变化,也就是环己醇亚硝酸酯在逐渐向环己醇转化,各个样品及标准品的转化程度又各不相同,因为每个样品和标准品不可能在同一时间进样,所以在处理完之后到进样前这段时间的长短不一,就造成了各个样品检测结果的误差。仔细研究了一下,标准中处理方法。衍生过程中是需要在冰浴中进行的,低温应该就是这个衍生反应的必备条件。但是在提取之后,提取液就离开了冰浴的环境,这个是否就是环己醇亚硝酸酯向环己醇转化的有力时机呢?为了解开这个疑问,我们又做了如下实验。我们在衍生之后,让其停留在比色管中,在进样之前一直停留在冰浴里面,这样的实验结果发现,转化的确是像被暂停了一般,因为不同时间进样的重复性非常好。实验结果让我们很兴奋,但是这种操作方法对我们自动进样器来说没有什么优势,不能完成序列进样,非常浪费时间。于是我们又进行了下一步试验。既然在比色管放在冰浴没有问题,是否可以把提取液转移到进样瓶中,再把进样瓶保存在冰浴里,看看是否变化呢?实验结果证明,我们的想法是正确的,只要控制低温环境就可以延缓这种转化。由此我们也总结出一个规律,这个转化其实是与温度高低有密切关系的。所以尽量降低环境温度也是一个不错的方法。这种转化似乎是无法彻底避免的,只能尽量去降低室温来延缓其转化速度,尤其是在夏天,空调温度设低一点。另外,我们也发现安捷伦气象色谱仪的百位盘有一个特殊的地方,我想应该就是可以接冷凝水或者别的降温措施,来提供低温环境的。http://ng1.17img.cn/bbsfiles/images/2014/12/201412292105_529965_2830723_3.jpg以上是我的一点拙见,希望对大家有所启示。进一步研究还需要大家继续进行,有不妥之处,欢迎大家拍砖。
谁可以告诉在哪里可以买到反-2-己烯醛、正丙醇、异丁醇、正丁醇、异戊醇、正己醇等物质的标准品啊最好是提供厂家的联系方式,谢谢了!!
氨基酸标准品哪里买?
我们有一个酯类API,合成过程中加入正己醇生成的酯,该API需要测定正己醇残留,正己醇沸点约157℃,因此采用150℃的顶空条件测定样品,发现样品中的正己醇残留色谱峰面积其大,可能是该API在顶空条件下降解生成了正己醇,那么这样我如何实验证明该正己醇是降解的,样品中的正己醇残留应该怎么测定呢?谁有过类似的经历,分享一下。
最近要用高效液相PITC柱后衍生检测食品中氨基酸含量,我看很多文献都有氨基酸标准品,但是有的用了18种,也有用17种,15种的,就像问一下各位大佬,这个是有什么标准关于检测那一类食品一定要检测哪几种氨基酸嘛
你们好,我想请教一下,用液相做氨基酸检测所需要用的标准品的纯度应该在多少以上啊?是不是用国家级标准物质会浪费?今天联系了很多供应商,想请购苏氨酸单独标准品,国家标准物质网上的苏氨酸对照品的价格在70元(100mg/瓶),标准物质750元/瓶(200mg/瓶),和别的供应商联系说中检所的标准品要600多元/瓶,纯度在99%,现在我就不确定了,到底标准品和标准物质是不是一样的,是不是检测的范围有所局限?请高人明示!谢谢
请问各位老师,有没有做过地毯胶黏剂检测?按照GB18587-2001,测试地毯胶黏剂中的2-乙基己醇,具体分析方法采用ISO16017-1:2000。请问是将胶黏剂涂抹在模拟板上后,按照小型环境试验舱进行采样,然后对样品分析吗?
求3-氨基-5正丙胺基-1,2,4-三氮唑标准品,CS号也不知道是多少
大家好,我一直用安捷伦1120LC做17种氨基酸的含量测定,但是在检测过程发现因检测氨基酸的量很低,买的安捷伦的标准有时会偏低,使测定结果波动很大,想请问各位,买哪里的氨基酸标准比较稳定准确?再者,在检测氨基酸怎样才能提高准确度?
做食品中的甜蜜素,标准是在坛墨买的,叫水中甜蜜素(问过客服,可以拿来做食品中甜蜜素),浓度是10000ug/ml,按标准配置6个浓度,和样品一起衍生,结果发现标准有很多杂峰(标准6即0.5mg/mL,见图一图二),图三图四是标准1(0.01mg/L),图五是正庚烷进了一针,想请教一下为什么标准里面会有这么多杂峰,目标产物该如何区分(杂峰太多,环己醇亚硝酸脂和环己醇区分不出)[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/05/202305111552079463_4369_5536399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/05/202305111552133668_9821_5536399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/05/202305111552497656_4144_5536399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/05/202305111553019934_4992_5536399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/05/202305111553517283_5497_5536399_3.png[/img]
化学位移试剂的原理是什么啊??怎么能将化学位移非常接近的H的信号给分开,能分开正己醇的所有的H吗??
《食品中氨基甲酸乙酯测定》国家食品安全标准终于制定完成发布,大家在使用过程有什么问题,可以联系我互相交流。







 我要推广仪器
我要推广仪器
 下载APP
下载APP