秋燥,容易出现咳嗽、咳痰等呼吸道不适症状。止咳药水由于功效好、口感不错成了许多人的首选。专家指出,止咳药水中的磷酸可待因会导致身体钙质流失!中成止咳药有可待因成分吗?迪马柱能检测止咳药水中的磷酸可待因?
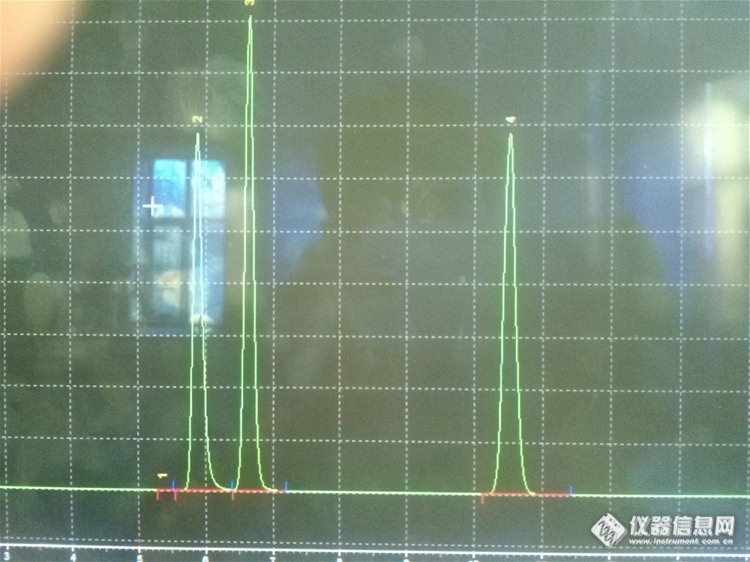
我昨天配了一个磷酸可待因的对照,处理过配置程是105度烘至恒重,然后用流动相(流动相是甲醇-0.03mol/L的磷酸氢二钠=20:80)配制。昨天进样后只有一个主峰,很正常,但是今天我又进了样,发现在主峰(15分钟出峰)前面出现一个比主峰更大的杂峰(5分钟左右)而且峰形很规则,我以为是流动相的溶剂峰,补进了一针流动相,但是在5分钟左右并为出峰,所以不是溶剂峰。请问下各位,那个杂峰是从哪儿来的呢,会不会是磷酸可待因和流动相发生了啥子化学反应,产生了一个新的东西呢?如果不是,那个杂峰又是从哪儿来的呢?
样品为复方维生素,其中一项是核黄素磷酸钠,没找到核黄素磷酸钠的对照品,故用的核黄素对照品样品制备:先用水溶,然后用流动相稀释,流动相弱酸性做出的结果比标示量低了很多啊用核黄素对照品代替核黄素磷酸钠对照品,请问结果可信吗?
[center]SFDA要求各地严管含可待因复方口服溶液[/center]针对近期陆续出现的滥用含可待因复方口服溶液的问题,国家食品药品监督管理局(SFDA)新闻发言人颜江瑛在12月8日召开的例行新闻发布会上说,SFDA近日下发了《关于加强含可待因复方口服溶液管理的通知》,对生产、批发、零售含可待因口服溶液提出了更为严格的管理措施。 首先,要开展含可待因复方口服溶液专项检查。各地食品药品监管部门要在各自辖区内组织开展一次针对含可待因复方口服溶液的专项检查,对辖区内含可待因复方口服溶液的经营情况进行一次全面检查,重点检查药品的生产、批发企业含可待因复方口服溶液的销售流向,以及药品零售企业是否严格凭处方销售含可待因口服溶液的情况。 其次,进一步规范含可待因复方口服溶液的经营行为。要求药品生产企业对销售客户进行全面清理,适度集中销售渠道,选择信誉和管理比较好的药品经营企业经营该品种,不得向无资质的单位或个人销售含可待因复方口服溶液;药品生产、批发企业销售时,必须严格执行《药品管理法》、《药品流通监督管理办法》等相关规定,核实购买方资质证明材料、采购人员的身份证明等情况,无误后方可销售,并跟踪核实药品到货情况;药品零售企业应当严格凭处方销售含可待因复方口服溶液。 再次,进一步加大对含可待因复方口服溶液生产经营违法违规行为的打击力度,对药品生产、经营企业未按规定销售的,要严格依照《药品管理法》、《药品流通监督管理办法》等规定处理;对药品批发企业将含可待因复方口服溶液销售给无资质的单位或个人以及药品零售企业未严格凭处方销售,造成滥用并导致严重后果的,一律吊销其《药品经营许可证》。 颜江瑛在发布会上介绍说,含可待因复方口服溶液临床主要用于镇咳,疗效较好,使用面广,是常用的镇咳药品。正常使用是安全有效的,但如果大剂量使用,其中所含的磷酸可待因和麻黄碱两种成分作用叠加,会产生致幻作用,极易造成滥用,对人体造成损害,甚至危及生命。上世纪90年代末,我国部分地区开始出现含可待因复方口服溶液滥用问题,滥用人群以青少年为主。自发现这一问题后,我国一直密切监测该类药品的滥用情况,2000年以来多次采取措施加强对该类药品的管理,2005年更明确规定药品零售企业必须严格凭处方销售含可待因复方口服溶液。 据悉,目前该类药品在我国共有13个品种获准上市,包括9个国产品种和4个进口品种。在国际上,也未对该类药品实行特殊管理。信息来源:医药经济报
视频参赛,吗啡可待因快检!
自制火锅底料中的罂粟碱、吗啡、那可丁、可待因和蒂巴因检测中吗啡标准品从哪里买啊?只是检测罂粟壳带入的成分吗?
《国食药监食 3 号/附件 3:火锅食品中罂粟碱、吗啡、那可丁、可待因和蒂巴因的测定》有谁能提供一下这个方法的文本?谢谢
可待因 蒂巴因 吗啡下游产品有哪些?医药或其他
开始是这样的,新配制的流动相(氯化钾和磷酸二氢钾混合液,PH 6.5),流速是0.8ml/min 新柱子,对照品为什么会变呢http://ng1.17img.cn/bbsfiles/images/2014/03/201403251443_494159_2793190_3.jpg
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
近日在做一个日本上市缓释制剂的仿制,对其释放度检查标准有些疑问,特别是其供试品的吸收度计算方法很特别,希望园里的老师给分析指点一下,谢谢!释放度的测定取本品,照释放度测定法,采用溶出度测定法第二装置,以Ph1.2盐酸溶液500ml为溶剂,转速为每分钟100转,依法操作,经1小时时,取溶液10ml,过滤,作为供试品溶液(1);弃去上述各容器中的酸液,加已预热至37±0.5℃的Ph7.5磷酸盐缓冲液500ml,继续运转至2小时时,取溶液10ml,过滤,作为供试品溶液(2);并补加同体积的Ph7.5磷酸盐缓冲液,继续运转至5小时时,取溶液10ml,过滤,作为供试品溶液(3);另取对照品约0.1g,精密称定,置50ml量瓶中,加甲醇适量使溶解,并稀释至刻度,摇匀;精密量取此溶液各1ml,置100ml量瓶中,分别加Ph1.2的盐酸溶液和Ph7.5磷酸盐缓冲液稀释至此刻度,摇匀,作为对照品溶液(1)和对照品溶液(2)。照分光光度法,供试品溶液(1)在360nm和450nm波长处测定吸收度,计算吸收度差值;其他各对照品溶液及供试品溶液均在360nm波长处测定吸收度,分别计算不同时间的释放量。
近日在做一个日本上市缓释制剂的仿制,对其释放度检查标准有些疑问,特别是其供试品的吸收度计算方法很特别,希望园里的老师给分析指点一下,谢谢!释放度的测定取本品,照释放度测定法,采用溶出度测定法第二装置,以Ph1.2盐酸溶液500ml为溶剂,转速为每分钟100转,依法操作,经1小时时,取溶液10ml,过滤,作为供试品溶液(1);弃去上述各容器中的酸液,加已预热至37±0.5℃的Ph7.5磷酸盐缓冲液500ml,继续运转至2小时时,取溶液10ml,过滤,作为供试品溶液(2);并补加同体积的Ph7.5磷酸盐缓冲液,继续运转至5小时时,取溶液10ml,过滤,作为供试品溶液(3);另取对照品约0.1g,精密称定,置50ml量瓶中,加甲醇适量使溶解,并稀释至刻度,摇匀;精密量取此溶液各1ml,置100ml量瓶中,分别加Ph1.2的盐酸溶液和Ph7.5磷酸盐缓冲液稀释至此刻度,摇匀,作为对照品溶液(1)和对照品溶液(2)。照分光光度法,供试品溶液(1)在360nm和450nm波长处测定吸收度,计算吸收度差值;其他各对照品溶液及供试品溶液均在360nm波长处测定吸收度,分别计算不同时间的释放量。
药典 金钱草含量测定以甲醇一O.4%磷酸溶液(50:50)为流动相;检测波长为360nm,温度30℃。 对照品溶液的制备 取槲皮素对照品、山柰素对照品适量,精密称定,加80%甲醇制成每1ml各含槲皮素4μg、山柰素20μg的溶液,即得 供试品溶液的制备 取本品粉末(过三号筛)约1.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,密塞,称定重量,加热回流1小时,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过。精密量取续滤液25ml,精密加入盐酸5ml,置90℃水浴中加热水解1小时,取出,迅速冷却,转移至50ml量瓶中,用80%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。 本品按干燥品计算,含槲皮素和山柰素的总量不得少于O.10%。
罂粟蒴果中可待因、吗啡、罂粟碱的GC-MS检验罂粟含有丰富的吗啡等吗啡型生物碱,为一年生草本,有乳状液体,茎自基部分枝,高60-120cm ,有白霜,无毛,下部叶有叶柄,上部叶抱茎,心脏形,有不对称粗齿,蒴果球形,直径2.5-5cm ,花期4-5月,果期6-8月。随着我国禁毒工作的不断深入,在铲除非法种植dupin原植物的同时,在法庭科学上,须对这类植物进行相关生物碱的检验。本文介绍了利用GC-MS对罂粟蒴果中可待因、吗啡、罂粟碱进行检测,方法简单。1 实验部分1.1仪器与试剂Varin3900 /2100GC-MS. 甲醇,丙酮,无水硫酸钠,均为分析纯。三种标准品均从公安部物证鉴定中心购得。1.2实验条件气相色谱条件:DB-5 30m X0.25mmX0.32 um 弹性石英毛细管柱;柱温:80℃(1min)20℃/min280℃(19min);进样口温度:250℃;分流比40:1;载气:高纯氦;进样量1ul.质谱条件:传输线温度240℃,离子阱温度150℃,EI源(70eV),扫描范围40-550amu.1.3检材的提取与净化取检材2-5克,加10克无水硫酸钠研磨,以丙酮提取,过滤,水浴或微波挥干,1ml甲醇溶解待检。2 结果与讨论该法中,罂粟蒴果中可待因、吗啡、罂粟碱的保留时间分别为12.33min、12.70min、16.26min,离子碎片峰依次为:可待因m/z299、162、124;吗啡285、162、215、124;罂粟碱339、324、308、292、266等。
大家好,我有一个样品是酸性的,对照品有点拖尾,但是不是很严重,样品比较严重,我加了点磷酸,没有得到改善!我不知道可以加点三乙胺吗?
求助:中药口服液质量标准制定的一些问题 第一次做新药,是一个中药复方口服液,主要成分中含有氢溴酸东蒗菪碱,质量标准制定如下: 5.1.1色谱条件 色谱柱:Promosil C18(250×4.6mm,5洀,博纳艾杰尔公司);流动相:0.07mol/L磷酸钠溶液 (用磷酸调 pH值至6.0,含0.0175mol/L十二烷基硫酸钠)-乙腈 (2:1);流速:0.8ml/min;检测波长:216nm;柱温:25℃;进样量:10ul。理论塔板数按氢溴酸东莨菪碱峰计算应不低于10000。 5.1.2 供试品和对照品溶液的制备 5.1.2.1 对照品溶液 精密称取氢溴酸东莨菪碱10.18mg,置10ml容量瓶中,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)使溶解,并稀释至刻度,摇匀,制成每1ml含1.018mg的溶液(东莨菪碱重量=氢溴酸东莨菪碱/1.445)。 5.1.2.2 供试品溶液 精密量取本品溶液5ml,置锥形瓶中,加入2mol/L盐酸溶液30ml,超声处理(功率250W,频率40kHz)30分钟,放冷,滤过,滤渣和滤器用 2mol/L盐酸溶液分数次洗涤,合并滤液和洗液,用浓氨试液调节pH值至9,用三氯甲烷振摇提取4次,每次30ml,合并三氯甲烷液,回收容剂至干,残渣用0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)溶解并移至5ml量瓶中,稀释至刻度,摇匀,即得。 5.1.3 阴性对照品溶液的制备 按处方比例及工艺,制备缺洋金花的阴性样品,再按供试品溶液的制备方法制备不含洋金花的阴性样品溶液。 5.1.4 测定方法 精密吸取上述对照品溶液与供试品溶液各10u1,分别注入液相色谱仪(自动进样仪),测定峰面积,以外标法计算含量。 5.1.5 系统适用性试验 分别吸取对照品溶液、供试品溶液及阴性品溶液10ul注入高效液相色谱仪,阴性对照品在氢溴酸东莨菪碱峰位置处无吸收峰,即其它成分对测定无干扰,氢溴酸东莨菪碱峰与其它组分峰基线分离良好。 5.1.6 线性试验 精密称取氢溴酸东莨菪碱对照品10.16mg,置25ml量瓶中,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)溶解并稀释至刻度,摇匀。精密吸取0.5、1.0、1.5、2.0、2.5、3.0ml,分别置10ml量瓶中,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)溶解并稀释至刻度,摇匀。按上述色谱条件测定,分别进样10ul,测得峰面积积分值。以对照品的浓度为横坐标,峰面积积分值为纵坐标,绘制标准曲线,得回归方程为:A=1.3×107C-9243.77,相关系数r:=0.9997。表明进样量在0.2032~1.2192ug范围内呈良好线性关系。 5.1.7 稳定性试验 精密量取供试品溶液5ml,按供试品(批号:2009010501)溶液制备方法制成供试品溶液,间隔一定时间(0,4,8,12,24,48h)重复进样,每次进样10ul,测得峰面积的RSD为1.78%,表明该溶液在48小时基本稳定。 5.1.8 精密度试验 精密吸取上述对照品溶液10ul,重复进样6次,测定东莨菪碱的峰面积,结果 RSD%为0.58%(n=6)。表明本方法精密度良好。 5.1.9 重复性试验 精密量取供试品溶液(批号:2009010501)5ml,按供试品溶液制备方法平行制得6份供试品溶液,分别测定东莨菪碱的含量,结果RSD为1.42%(n=6)。表明本方法有较好的重复性 。 5.1.10 加样回收率试验 5.1.10.1 对照品溶液的制备 精密称取氢溴酸东莨菪碱对照品约7.8mg,置100ml的容量瓶内,加0.07mol/L磷酸钠溶液(用磷酸调pH至6.0)使溶解,并稀释至刻度,即得。 5.1.10.2 采用加样回收率测定方法,精密量取已知含量的样品2.5ml,精密加入上述对照品溶液2.5ml/2ml/3ml,按供试品溶液制备方法平行制得6份供试品溶液,依法测定,结果平均回收率为106.7%,RSD为1.42%。 回收率=(测的东莨菪碱含量—样品中东莨菪碱含量)/加入东莨菪碱量 因为是第一次作含量测定,有许多不明白之处,中药质量标制定准指导原则也说得不清不楚,许多都是按照自己理解摸索来的。 通过测定样品的平均含量为0.077mg/ml。线性试验是根据样品的平均含量,以样品的平均含量为中间值,制定的线性取样范围。 根据加样回收中对照品加入量与样品含量之和在线性范围的原则,按照80%,100%,120%的原则加入对照品,但是结果很不理想,加样回收率超过了200%,不知道是什么方面的原因,很着急,请大家帮忙分析一下,找出有错误的地方,我马上更改。
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。
因为刚接触药品检验,在做核黄素磷酸钠的游离磷酸的测定的时候,做了几次都不合格。 按照标准要求进行对照及样品配制,对照品为蓝色的澄清液体,但是样品很浑浊,土黄色。在730nm处进行测定,结果不符合规定。 因为样品很浑浊,所以吸收值很大(2.0),大过了对照品; 我们把样品用0.45的滤膜过滤,吸收值(0.35)低于对照,但是我们怀疑过滤的影响太大,我们就再次过滤样品,吸收值又降低了一半(0.13). 请各位大侠帮帮忙啊!!!
对照品4个,第2(延胡索乙素)、3(盐酸小檗碱)个色谱峰前出现了小峰,不知道什么原因引起的,之前用相同色谱条件跑过,也没有小峰出现,现在连续进了五针都是主峰前有小峰。请各位帮我分析分析!流动相是0.1%的磷酸水:乙腈,对照品用甲醇溶解。色谱条件如图[img=,690,284]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044560750_5451_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044563666_9373_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044551717_7304_5351399_3.png[/img][img=,690,618]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044568830_7416_5351399_3.png[/img]
[color=#444444]请问大神们,麻烦大家帮我分析一下,我实在没法了:我用浓度为30微克/ml的山奈素对照品溶液跑高液,色谱条件是甲醇:0.4%磷酸水比例50:50,,流速1ml/min,柱温30℃,检测波长为360nm.进样量为20微升.为什么跑不出来任何峰形啊。。用异鼠李素对照品都能跑出来的,山奈素的出峰位置应该是在异鼠李素之前的。。。[/color][color=#444444][/color]
因为刚接触药品检验,在做核黄素磷酸钠的游离磷酸的测定的时候,做了几次都不合格。 按照标准要求进行对照及样品配制,对照品为蓝色的澄清液体,但是样品很浑浊,土黄色。在730nm处进行测定,结果不符合规定。 因为样品很浑浊,所以吸收值很大(2.0),大过了对照品; 我们把样品用0.45的滤膜过滤,吸收值(0.35)低于对照,但是我们怀疑过滤的影响太大,我们就再次过滤样品,吸收值又降低了一半(0.13). 请各位大侠帮帮忙啊!!!
乙腈-0.1%磷酸测柠檬苦素成分含量,前几个月一直是单峰,今天做出峰时间一样,但是峰裂成两个了流动相没有变动,对照品一开始怀疑变质了重新配了,针也洗了,新配的对照品出峰还是裂开的,这种情况只可能是柱子塌陷了吗?这根柱子没法再用了吗?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2021/03/202103242018018319_7823_5205322_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2021/03/202103242018023739_744_5205322_3.png[/img]
[color=#333333]对照品与标准品概念[/color][color=#333333]对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质,而标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示.文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已[1,2],造成错误的原因,可能是有的药品既有对照品,又有标准品.例如,当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品.即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的.[/color]
对照品:用于鉴别、检查、含量测定和校正检定仪器性能的标准物质;对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。 标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。 对照品与标准品概念不清?对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已,造成错误的原因,可能是有的药品既有对照品,又有标准品。 例如:当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品。即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的。
仪器:LC-100 二元高压,Exformma 经济型C18色谱柱,5µm,4.6×250 mm,加保护柱。流动相:乙腈-O.05mol/L磷酸二氢钠溶液(50:50);波长为440 nm;柱温40℃,进样量20μl。理论板数按血竭素峰计算应不低于4000。对照品溶液的制备 取血竭素高氯酸盐对照品9mg,精密称定,置50ml棕色量瓶中,加3%磷酸甲醇溶液使溶解,并稀释至刻度,摇匀,精密量取1ml,置5ml棕色量瓶中,加甲醇至刻度,摇匀.即得(血竭素重量=血竭素高氯酸盐重量/1.377)。供试品溶液的制备取本品适量,研细,取O.05g,精密称定,置具塞试管中,精密加入3%磷酸甲醇溶液10ml,密塞。振摇3分钟,滤过,精密量取续滤液1ml,置5ml棕色量瓶中,加甲醇至刻度.摇匀,即得。血竭素的保留时间为6.4,但拖尾因子为3 ,不符合中国药典规定,请教大家如何处理?
今天国家局的专家们对我们进行工艺核查,问我们对照品的领用问题,一开口对照品有效期是多长?(不涉及特殊品种)未用完的对照品要不要进行再标定?多长时间标定?怎么保证未领用已开口的对照品的含量?企业对照品管理办法是否经过验证?
正在做一个6类化药,对照品中检院有提供;为了省钱,在中检院购买了一支对照品(100mg ),用来标定购买的原料药(GMP厂家提供)作为工作对照品;标定方法:按原料药质量标准全检,HPLC外标法标定含量(没做结构鉴定); 已经使用工作对照品做完了含量和溶出度的方法验证;问题来了: 1.在中检院可以提供对照品的情况下,可以使用自己标定的工作对照品么?(CDE审评三部张哲峰的文章:“不应使用其他来源者”,这话的意思是必须在中检院购买?); 2.如果可以使用工作对照品,那么,这样简单标定就可以使用了么? 3.如果不能使用工作对照品,那现在的情况下怎么补救? 请各位指教,谢谢!
[font=宋体]实验室新配置了对照品,对于对照品这方面的管理体系目前是没有的,现在有个疑问是实验室配制的对照品还需不需要使用部门去验收,制定期间核查计划的话,是新配一批就更新补充计划,还是可以笼统的写,不体现批号,一年只写一次?[/font]
各位大神,请教一个小白的问题,关于HPLC中对照品的配制。外标法测定麦芽糖浓度。麦芽糖对照品是99%纯度的一水合物,请教问题是:真空干燥24h后,对照品中的一水合物的水是否会被去除掉,称量的时候是按一水合物的分子量计算来称量,还是按没有水的纯麦芽糖分子量来称量比如要求称1g就在天平称1g?
标准对照品有没有出问题的,做了多年的能力验证了,是不是遇到过对照品的问题呢???







 我要推广仪器
我要推广仪器
 下载APP
下载APP