自制杂质对照品,新药申报时杂质含量方面需要做哪些工作?按照CTD格式要求,对于自制对照品应该“简述含量和纯度标定的方法及结果”,具体做应该怎么做?请各位专家指导,谢谢!
相关物质检测的时候通常会用到杂质对照品,关于这个杂质对照品你是如何管理的?对它的含量与纯度有木有做过分析检测?有效期是怎么规定的?
如题,请知道的朋友告诉我,先谢谢了! 我即将参与做的一个3.1类新药项目,其杂质定量研究正需要考虑这个响应因子的问题,以前做的都是补充申请,还没有考虑响应因子不一致的情况,现在考虑了一个方案,那就是液相色谱检测,液相色谱和紫外同步进行,如果我们这个新药项目杂质可得,并且有对照品,那么就可以绘制杂质和对照品的标准曲线,杂质标准曲线的斜率除以对照品标准曲线的斜率的比值就是响应因子吧?我想参照指导原则确定到底什么范围时可以用主成分的自身对照法计算含量,什么范围时,宜用杂质对照品法计算含量,也可用加校正因子的主成分自身对照法?您有好的建议吗?期待您的帮助。
一直很纠结,杂质对照品的稳定性需要做吗?我曾请教过别人说是不用作,可是如果不做,当作为对照品时,无论结构还是浓度一旦发生变化,岂不是杂质的检测就受影响吗?
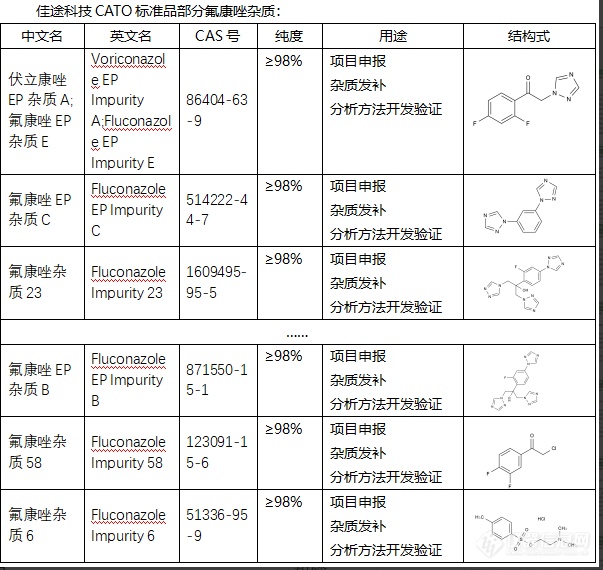
氟康唑杂质可能来自原料药、副反应产物或分解产物,以及其在存储过程中可能发生的变化。这些杂质如果未被及时检测和控制,可能对药物的效力和安全性产生影响,包括药效降低、不良反应增加等。因此,检测和控制氟康唑中的杂质是药品生产过程中的重要环节,对药物质量、安全性以及疗效的保证至关重要。研究分析这些杂质,还可以优化生产工艺,减少杂质产生,提高药物的质量和疗效。CATO标准品对沙格列汀杂质的研究也能帮助优化制药流程,找出产生过多杂质的环节,从而改进工艺,提高药品的质量和疗效。[img=,603,570]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052101491000_1515_6381668_3.png!w603x570.jpg[/img]
丙谷二肽杂质对照品名称 环-(L-丙氨酰-L-谷氨酰胺) 规格:0.25gL-焦谷氨酰-L-丙氨酸 规格1g"N-(2)-D-丙氨酰-L-谷氨酰胺" 规格:0.25gL-丙氨酰-L-谷氨酸 规格:1gL-焦谷氨酸 规格:125mg
请教专家:如何用HPLC准确定量样品中的一个杂质,目前有样品、杂质对照品,样品中的杂质已用百分比法测出。如用外标法,溶剂及波长是否要一致。谢谢!
液相分析时,对照浓度一般是多少,根据什么来定?杂质对照呢?
[color=#DC143C]请问大家在用TLC测定有关物质的时候,有没有遇到过把供试品溶液稀释N倍后作为对照溶液的情况?我很疑惑:供试品溶液的浓度比对照溶液深,为什么杂质斑点不得比对照溶液深呢?这样的对照能说明什么问题呢?谢谢![/color]我是在中国药典上见到的这种方法:乙胺嘧啶的有关物质(05版CP第3页):取本品,加三氯甲烷-甲醇(9:1)制成每1ml中含20mg的溶液,作为供试品溶液;精密量取适量,加同一溶剂稀释成每1ml中含50ug的溶液,作为对照溶液。照薄层色谱法检验,……供试品溶液如显杂质斑点,与对照溶液的主斑点比较,不得更深。
[font=宋体]1.用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做对照品的分子量的确认(scan模式),[/font][font='Times New Roman',serif] [img=,159,138]file:///C:/Users/ADMINI~1/AppData/Local/Temp/msohtmlclip1/01/clip_image002.png[/img][/font][font=宋体]杂质[/font][font='Times New Roman',serif]-B [/font][font=宋体]测得[/font][font='Times New Roman',serif]m/z[/font][font=宋体]是[/font][font='Times New Roman',serif]314.0[/font][font=宋体](最高响应峰)[/font][font=宋体],与实际相差[/font]17[font=宋体],而本该有的[/font][font='Times New Roman',serif]m/z331[/font][font=宋体]的峰很小[/font]2.[font=宋体]用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做对照品的分子量的确认(scan模式),[/font][font='Times New Roman',serif][img=,136,110]file:///C:/Users/ADMINI~1/AppData/Local/Temp/msohtmlclip1/01/clip_image002.png[/img][/font][font=宋体]杂质[/font][font='Times New Roman',serif]-L [/font][font=宋体]测得[/font][font='Times New Roman',serif]316.1、347.1(两个响应高的峰),[font=&]347.1是对的上,[font=&]316.1不知道怎么产生的?[/font][/font][/font]
我公司求购下列对照品:USP Gabapentin Related Compound D RS [(1-(3-oxo-2-aza-spiro[4.5]dec-2-ylmethyl)-cyclohexyl)-acetic acid] (C18H29NO3 307.43) . 1S (USP29)联系方式: wdzhang@du-hope.com
现有一原料药,已知其杂质为abcde,并有相应的杂质对照品,请教大家怎么测定杂质的校正因子?
普拉克索杂质A,B,C,D,E欧洲药典标准。进口注册标准中代码【BI-II751XX】 【BI-II786BS】 【BI-II820BS】BI-II 546 CL】常用杂质对照品
借贵版人气问一个问题:测定样品中的杂质时,什么情况下不用杂质标准品做对照,而采取把样品稀释后做自身对照?能给出自己工作中具体例子的,给悬赏分。谢谢!我说的是药品中的有关物质(生产过程中带来的有机杂质),无机杂质和残留溶剂不算。有关物质包括已知杂质和未知杂质,二者加起来是总杂质。我在中国药典上见到过这样的方法:乙胺嘧啶的有关物质(05版CP第3页):取本品,加三氯甲烷-甲醇(9:1)制成每1ml中含20mg的溶液,作为供试品溶液;精密量取适量,加同一溶剂稀释成每1ml中含50ug的溶液,作为对照溶液。照薄层色谱法检验,……供试品溶液如显杂质斑点,与对照溶液的主斑点比较,不得更深。详见下面的帖子:http://www.instrument.com.cn/bbs/shtml/20070831/964769/[color=#DC143C]我在5楼举了几种情况,都是我翻译资料时遇到的。[/color]药品审评中心的老师也发过相关的讨论,见下:[color=#00008B]关于HPLC主成分自身对照法检查有关物质时检测波长确定的讨论[/color]http://www.cde.org.cn/page/framelimit.cbs?ResName=dzkw据说这种稀释后自身对照的方法应用得挺多,但[color=#DC143C]我不知道为什么要这样用。[/color]
请教下各位,一般做溶出曲线时,一个介质配一份该介质下的对照;但是遇到多个介质一起做的时候,是否可以共用一份对照?假如,同时配制的多介质对照品f值只相差2%以内,是否可以认为这几个介质的对照可以共用呢?就不再需要每个介质单独配对照?
采用面积归一化法测定高分子杂质,但系统适用性的对照品溶液峰面积rsd达到9%以上,这样会影响杂质定量么,有人说峰面积归一化法不用对照品峰面积,不用管,有做过的么?(对照品溶液和杂质不是一个物质)
[B]杂质对钛白粉有何影响及提高其白度的方法[/B] 对于颜料钛白粉,除了生产上采取一些措施来提高白度以外,关键还在于杂质的去除,杂质对白度的不良影响是很大的,杂质去除得越彻底,产品白度就越高,这对用在涂料中的意义就更大。 在钛白粉生产过程中,如果杂质去除不彻底,当用高温煅烧时,很多杂质元素如铁、锰、钒、铅、铬、钴、铈铜、镉、镍、钼等以氧化物状态存在,这些带色的氧化物就表现出各种色相,使整个钛白粉的色相受到影响而不纯白,以致大大影响了产品的质量。因此,必须采用多种方法除去杂质。 1选矿除杂质 要除去杂质,首先就要选矿。因为任何钛铁矿一般都混杂有不少脉石和共生、伴生、复合的其它矿物。选矿就是利用矿物不同的物理化学性质,采用各种有效方法,将钛铁矿与它们分离。例如摇床的重力选矿,可以除去铸铁矿中的大部分脉石,再用磁选机进行磁选,让矿物通过磁场,由于钛铁矿是导磁率高、能被磁铁吸引而本身不能吸铁、可磁化又可去磁的顺磁性矿物,而能磁盘吸引,其它导磁率低的非钛铁矿,不能被磁盘吸引而得到分离。 2除不溶性杂质 硫酸法生产钛白粉,由酸解浸取得到的是浑浊不清的钛液,其不溶性杂质主要是颗粒较大的机械杂质和颗粒较小的胶体杂质。机械杂质是未起反应的钛铁矿物,属于粗分散状态,很容易沉析下来;胶体杂质主要是硅酸铝酸盐等,由于颗粒小,吸附H而带有相同的正电荷,由于同种电荷相斥,胶粒很难接近成比较大的颗粒而沉淀下来,因而具有较高的稳定性。解决的办法是用带负电荷的改性的聚丙烯酰胺胶体进行电性中和,使胶体粒子凝聚沉降而除去。但是由于胶体沉降不完全,经过硫酸亚铁的过滤后,仍有一些穿滤而存在于钛液中,必须用带有木炭粉为助滤层的板框压滤机进行压滤,直到检测滤液的澄清度合格为止. 3除铁杂质 在钛铁矿中,非钛杂质最多的是铁,并以二价和三价两种状态存在。将钛铁矿与硫酸作用,即生成FeSO和(SO)。由于FeSO只有在pH值大于6.5时才开始水解,因此在钛液水解过程中,因钛液的酸性较大,FeSO就始终保持溶解状态,在偏钛酸洗涤时得以除去。而Fe(SO)在pH值为1.7的酸性溶液中即开始水解生成Fe(OH)沉淀,其混杂在偏钛酸中,煅烧时即生成红棕色的FeO而使成品不够纯白。所以应用铁屑把Fe(SO)还原为FeSO。为了保证钛液中的三价铁全部还原为二价铁,还原反应还应略为过度,此时钛液中就有小部分四价钛还原为三价钛。三价钛的存在就可保证三价铁还原完全,可避免三价铁水解生成Fe(OH)进而影响产品白度。经过还原,钛液中全部是FeSO,此时冷冻钛液,Fe即达到过饱和状态而大量结晶析出,过滤即可除去大部分铁钛液中剩下的未结晶的FeSO,待水解生成偏钛酸进行水洗时,用水洗除去。由于滤饼的FeO质量分数超过90×10时,产品白度将受到影响。所以可采取漂白措施使FeO质量分数降低到30×10,并进一步除去痕量的钒、铬、铜等杂质。
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]
溶剂残留测定,对照品杂质峰是否应该删除好友回复:不应该大家说说看http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_670748_3111590_3.jpg
硅胶中含有的少量金属杂质对色谱峰有影响吗?如有,应该如何处理?
实验室研发的新产品即将生产,其中有一个[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]监控的步骤,需要用简单的方式来定量控制该物质的含量。杂质是异丙醚,此步是中间控制,需要将该杂质含量控制在1%以内,领导要求开发一个简便的方式来定量从而控制其含量。我想了一下,能否将产品与杂质1:1配制,溶解至100ml,那么此时杂质与产品的含量比即为杂质与产品的峰面积比。问过研发人员,中间产物中,产品大致的量他们是知道的,也就是产品的量已知。那么能否通过直接进样中间产物,观察产品与杂质的比值来判断杂质的大致含量呢?这个方式我在液相的杂质含量中其实有大致了解,但是无奈理解能力有限,其实没有太懂[b][color=#000000]加校正因子主成分自身对照法[/color][/b]与[color=#000000][b]不加校正因子的主成分自身对照法[/b]的[/color]计算方式。[b][color=#ff0000]举个例子:[/color][/b]第一种[font=&][color=#000000][size=14px][b]加校正因子主成分自身对照法[/b][/size][/color][/font]:假设我现在有杂质对照品,和产品对照品,我将两种配制成浓度已知的混合液,进样,已知校正因子(f)=(S杂质*C杂质)/(S产品*C产品)计算出校正因子,假设(f)=杂质:产品=1.2那么我现在有个中控液进样了,杂质:产品峰面积为1:10,产品在混合液中的含量差不多为50%,那么杂质含量占多少呢?杂质含量此时是6%吗?第二种[b]不[/b][font=&][size=14px][color=#000000][b]加校正因子主成分自身对照法[/b][/color][/size][/font]:这种我是完全没懂了,,,懵圈了。如果我第一种方法的理解没错的话,那么此次[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]中控方式能否用这种方式来计算杂质的大致含量呢。求教。
主成分的响应因子比已知杂质的响应因子小,2者相差较大,如何定量呢?已知杂质 Response Factoru/mL 5 11.5 10 12.3 30 23.9 50 31.8 80 36.5 100 36.4 主成分 5000 mcg/mLmcg/mL Peak Area 28679 Resp Factor 5.7 现有的方法,是用已知杂质对照品,来定量样品中的杂质含量。。响应因子相差较大,是否造成计算出的结果偏小??
有没有做过维生素A杂质的液相色谱分析?维生素A及其杂质维生素A环氧化物、维生素A醛、维生素A酸没有杂质对照品,以上物质在甲醇-水(90:10)流动相,C18*250mm柱能不能分的开?
气相色谱手动进样测杂质含量用主成分自身对照法准确吗,因为主成分自身对照法是比较供试品中杂质的峰与此对照溶液主峰的峰面积大小,那怎样确保供试品溶液和对照溶液前后两针的进样量一致呢?
[font=宋体][font=宋体]原料药,杂质含量测定,内标法,分析方法[/font][font=Calibri]---[/font][font=宋体]准确度。[/font][/font][font=宋体]1. [/font][font=Calibri]6[/font][font=宋体]份样品测定或[/font][font=Calibri]3[/font][font=宋体]种浓度各进[/font][font=Calibri]3[/font][font=宋体]针,再[/font][font=Calibri]5针[/font][font=宋体]对照品溶液求校正因子,得出杂质含量,求回收率[/font][font=宋体]或者[/font][font=宋体][font=宋体]2 向原料药样品加入已知杂质对照品,进样:[/font][font=宋体]①[/font][font=Calibri]5[/font][font=宋体]针对照品求校正因子,②样品溶液,③加入对照品的样品溶液。[/font][/font]是这样的吗?
1 准确度可由精密度,线性和专属性推算出来,是不是这三项合格,准确度必定合格,没必要算;或者算的话,怎么计算呢?2准确度应该用样品做还是标准品?2.1可向原料药中加入已知量杂质对照品进行测定;我做的是内标法,然后配6个100%的标准品溶液,或者80% 100% 120%,,是不是这样进样,:①5进针对照品溶液算校正因子,②进样品溶液,②进加对照品的样品溶液。
比如有关物质定量时,没有杂质对照品,需要自制,那自制的对照品都要测试些什么项目呢?先把我知道的写出来,求各位补充1、结构鉴定相关内容(UV,IR,H-NMR,C-NMR或更多,MS(是否必须做高分辨呢?))2、自制对照品纯度或含量测定(色谱纯度(HPLC或GC等),水分残留,有机溶剂残留,重金属残留等,最后的含量=色谱纯度-水分残留-有机溶剂残留-重金属残留等)还有其它的要做吗?
请问农药残留分析样品中的杂质对分析和检测会有哪些影响?
[size=3]请教一个计算,不胜感激!一个复方的制剂,用hplc的方法,检测一个已知有关物质的限度(不得过0.01%)。[/size][size=3]取5片量制得供试液,取已知杂质制得浓度为50微克每毫升的对照液。[/size][size=3]在供试液中检测到对应的已知杂质的杂峰,那公式是不是:[/size][size=3]A(样品中杂峰面积)*C(杂质对照浓度)/A(杂志对照峰面积)/W(5片粉质量)*100%请各位大侠指教,不胜感激![/size]
有关物质的含量测定时,有杂质对照品法,不加校正因子的主成分自身对照测定法,加校正因子的主成分自身对照测定法,本人一直不明白的地方是:加校正因子的主成分自身对照测定法有什么用呢?求算校正因子就得有杂质对照品,那既然有了杂质对照品,为什么不直接用杂质对照品法而非要拐弯抹角地用加校正因子的主成分自身对照测定法呢?求解……







 我要推广仪器
我要推广仪器
 下载APP
下载APP