在做二羟基丙酮的硅烷化分析的时候,出现一个问题:当标准样品的量比较少的时候比如10mg左右的时候,气相上出来的是二羟基丙酮的硅烷化的峰,但是当标品的来那个超过20mg的时候会出来二羟基丙酮硅烷化峰以及二羟基丙酮二聚体硅烷化峰,不知哪位专家做过此物质的分析,可否指点一下。
[color=#444444]我需要用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做羟基丙酮的外标线。但是羟基丙酮溶液是放在饱和碳酸钠里面的,而钠会对极性柱造成影响吧。想问该如何解决这个问题。[/color]
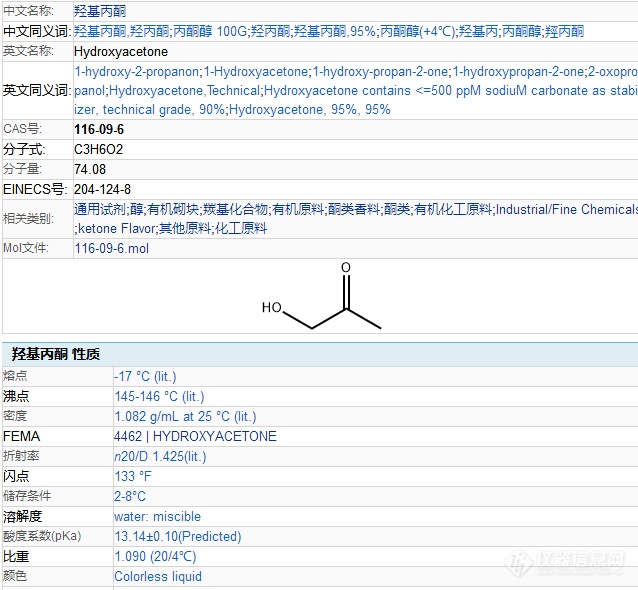
大家在分析食用香精时,是否经常看到有羟基丙酮这个物质,这不是直接加入的吧?[img=,638,590]https://ng1.17img.cn/bbsfiles/images/2021/10/202110090914118356_1121_2970225_3.png!w638x590.jpg[/img]
用溶剂解吸法测定工作场所空气中的苯、丙酮等有害物质时,用到的苯、丙酮等GCS级的标准物质都是低沸点,挥发性强的化合物,一般密封在安瓿瓶中,取用后,不知如何保存?我用过一瓶2mL的苯,敲开安瓿瓶,取完样后,用棉花堵住口子,再包上生料带,放在0~5℃的冰箱中保存,可过了约半个月,发现安瓿瓶中的苯全部挥发完了,不知大家如何保存?
您好!我正在做1,3-二氯丙酮水解生成1,3-二羟基丙酮的实验,具体的方法是1,3-二氯丙酮在乙醇保护羰基的条件下加碱冰浴水解完后再加入盐酸中和,我用液相色谱分析过生成物,色谱条件为C18的的柱子,柱温25度,波段200,流动相甲醇(80%)的水溶液,检测后再2.5min有连续的峰出现,我想知道怎样能把这些峰分开,您能指导一下对于这样的物质我该用什么条件来进行分析么?我也单独分析过1,3-而羟基丙酮的纯品,在2.5min时有峰,峰面积占比96.7%。
请教在甘油氧化制备二羟基丙酮的反应中使用哪种液相色谱柱分析效果较好,我们使用了氨基柱,效果不理想,求指点。
求助:对羟基苯甲酸丁酯钠和对羟基苯甲酸丙酯钠的相关行业标准,国内、国外的标准都可以。
各位前辈,请问以二硫化碳为溶剂的100ppm和10000ppm的丙酮标准溶液保存期一般为多少?其它如甲苯等挥发性有机物的标准溶液保存期一般又为多少?谢谢[em0808]
丙酮不溶物标准.rar[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=16338]丙酮不溶物标准.rar[/url]
请问一下买的丙酮中的环氧氯丙烷标准品,可以用丙酮做溶剂稀释吗,要用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]操作,标准里是用二氯甲烷做溶剂
乙酰丙酮烯醇式羟基氢的吸收峰为何不是尖峰,而是宽峰?
单位准备开展工作场所空气中丙酮的测定,采用的是二硫化碳溶剂解析方法,所用到的二硫化碳中丙酮标准溶液哪里可以买得到,浓度时多少,国家标准物质中心没有查到。
请问:农药标准储备液溶剂是丙酮,能否用移液器吸标准储备液来配标准曲线?移液器枪头会不会被丙酮溶解。
食品中丙酮酸的测定,标准曲线一直做的不太好,不知道有没有跟标准曲线对应的计算公式?比如说,我称取了0.6g样品,定容100ml,吸取10ml溶液,加1mol/L的HCl 20ml后,水浴3hrs,经萃取后测定吸光度为0.6,怎么来计算最终的丙酮酸含量?有直接应用的公式吗?谢谢!
[size=4]请教一下各位:食品中丙酮酸的测定,标准曲线一直做的不太好,不知道有没有跟标准曲线对应的计算公式?[/size][size=4]比如说,我称取了0.6g样品,定容100ml,吸取10ml溶液,加1mol/L的HCl 20ml后,水浴3hrs,经萃取后测定吸光度为0.6,怎么来计算最终的丙酮酸含量?有直接应用的公式吗?谢谢![/size]
丙酮的作用除了提取样品,做曲线按标准来说好像作用不大,直接省略只用二氯甲烷做稀释溶剂应该问题不大
现在做一个检测试验,要求试剂环己烷、丙酮、苯都要重蒸,可是不明白怎么进行重蒸。疑惑有几点:已购买的试剂都是分析纯的,还有重新蒸馏吗?即使需要重新蒸馏,需要加什么进行纯化哦?网上我查到两个苯、丙酮的纯化,但是环己烷怎么纯化,谢谢了
如何区分丙酮和甲苯?请教各位高手,我是一个刚接触气相色谱这方面的实验的一个新手,当我做丙酮和甲苯混合样时发现,这两个样品的峰不能区分出来,我已经试过很多方法了,但还是不可以,求高手指点。。。万分感激
对羟基苯甲酸甲乙丙酯的标准溶液有很多杂峰正常吗?我感觉理论上应该是只有三个大的色谱峰,结果出了十多个[img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310191022364344_1417_5979722_3.png[/img]
职业卫生丙酮标准溶液大家都是自己配制吗?解析效率都怎么做的啊?往活性炭管里面加色谱纯的丙酮吗?具体加多少怎么计算?
我用丙酮做为溶剂,做苯酐的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析,发现丙酮峰拖尾很严重,经常将苯酐的成分峰覆盖了,分析数据不准确,什么原因?望指导!我用的是KB-5的色谱柱
专家您好:我想用紫外分光光度计测丙酮的纯度,手上只有工业用甲醇与分析纯的丙酮,我用它们作出了标准工作曲线,但当我测工业丙酮含量时结果达到130%,麻烦您给予解答.
小弟最近在做乙酰丙酮分光光度法分析甲醛 按照GB/T 5009.49-2003 发酵酒卫生标准的分析方法做的。但是由于本人是第一次做,不知道标准曲线具体是什么样的,数据貌似不太对头。0.00 , 0.50 , 1.00 , 2.00 , 300 , 4.00 , 8.00 mL , l μg/mL的甲醛标准溶液于25mL比色管中,加水至10mL各加人2mL乙酰丙酮溶液,摇匀后在沸水浴中加热10 min ,取出冷却,于分光光度计波长420nm 处测定吸光度,绘制标准曲线。居然甲醛越高吸光度越低。而且做了好几次都是这样的!请求指点。哪位前辈也有做过这个标准曲线的发给我看看。谢谢! humangest@163.com
电极沥青甲苯不溶物的测定(ISO6376-1980)为什么用甲苯洗涤过滤液后还要用丙酮洗涤?丙酮洗涤是什么目的?操作过程称取沥青试样约1克,准至0.001g,测定前于105-110℃电路烘箱内加热过滤坩埚一小时,将其放入干燥器内冷却至室温,并称重准至0.001g。把试样放入锥形瓶中。加入100mL热甲苯(大约80℃)并摇动锥形瓶以溶解试样。注:为了防止因为软质沥青成型而造成在热甲苯中难以溶解,当沥青还是液体时移入锥形瓶是非常必要的。把冷凝器安装在锥形瓶上并打开循环。使甲苯稳定回流30分钟。停止加热并移开冷凝器。使用微弱的抽滤,立即用干燥且称重过的过滤坩埚过滤锥形瓶内液体。大约10毫升80℃的热甲苯洗涤锥形瓶并过滤洗液,过滤完成后,再用10mL热甲苯重复以上洗涤及过滤操作,直到所有锥形瓶内残渣都转移到过滤坩埚且使滤液无色。用大约10mL丙酮洗涤过滤坩埚,洗完以后,再用10mL丙酮重复以上操作。处理过滤坩埚:将过滤坩埚置入105-110℃恒温干燥箱内大约一小时,然后将其放入干燥器内冷却至室温并称准至0.001g。
请问各位大虾,谁有乙酰丙酮分光光度法分析甲醛的标准曲线吗?很急,希望各位给我个标准曲线,我第一次用这个方法,不知道标准曲线是什么样的?谢谢
我测丙酮(99%)里面的微量苯(0.01ppm),GC2010,FID,HP-5,30m*0.32mm*0.25um,柱温70度,奔最低检测限只能到0.5ppm,去别的公司看了,他们用顶空进样,安捷伦的7890A,康林的VOC专用柱,60m*0.32mm*1.8um,他们能做到0.1ppm。我不理解为什么要顶空进样(丙酮沸点56度,苯80度),普通进样方式不可以吗,如果我不用顶空进样,换和他一个类型的柱子是不是也能做到0.1ppm。谢谢
新手小白,刚接触GC-MS/MS,我用的是安捷伦7890A-7000B,柱子是DB-5的柱子,走的分别是羟基苯甲酸丁酯和2-氨基苯乙酮的标品,都是1ppm的,溶剂都是丙酮,升温条件50℃——220℃(15℃/min),不分流,然后不出峰。请各位大神多多指导!!!
农残的标准溶液必须用丙酮进行稀释吗?
甘油氧化产物 甘油酸 甘油 甘油醛 二羟基丙酮 乳酸如何分啊 文献里写的ALLTECH有机酸住 不详细啊
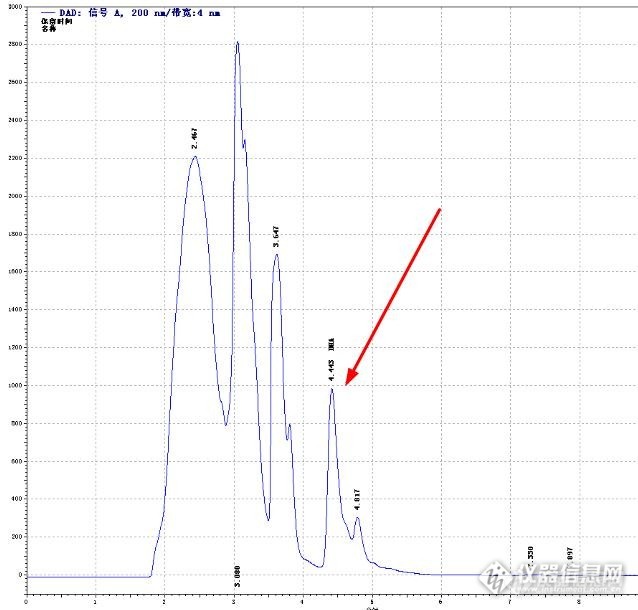
用氨基柱做二羟基丙酮(DHA),样品是合成出来的,直接进样,起初分离度非常好,可做了不到两个月分离度就不好了,究竟是什么原因呢?很纠结!流动相:乙腈:TFA(ph=3)=75:25流速:1ml/min检测器:200nm柱温:35℃正常谱图http://ng1.17img.cn/bbsfiles/images/2014/03/201403041528_491813_2563834_3.jpg非正常谱图http://ng1.17img.cn/bbsfiles/images/2014/03/201403041528_491814_2563834_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP