恶喹酸标准品用什么溶解啊?我们用的甲醇,都没溶!如果用氢氧化钠助溶还能用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测吗?
恶喹酸标准品用什么溶解啊?我们用的甲醇,都没溶!看网上说加氢氧化钠,但是以后怎么用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测呢?
求助:在哪能买到环丙烯酸(又叫锦葵酸)标准品。
按GB/T 20751-2006鳗鱼及制品中十五种喹诺酮类药物残留的测定 液相色谱-串联质谱法,标准规定10mg恶喹酸溶于100mL甲醇,制成100mg/L的标准贮备溶液,但实际上10mg恶喹酸不能完全溶于100mL甲醇,请赐教,有什么好办法?
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析脂肪酸甲酯标准品,怎样处理标准品?用什么溶剂稀释?
进口兽药质量标准硫酸头孢喹肟注射液Liusuan Toubaokuiwo ZhusheyeCefquinome sulfate Injection本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。【性状】 本品为类白色至浅褐色混悬液体;久置分层。【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。(2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。【检查】有关物质 照含量测定项下的方法。取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。装量 按最低装量检查法(附录67页)检查,应符合规定。【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。
急需动物性食品中噁喹酸和氟甲喹残留检测方法(鱼)--高效液相色谱法的检测标准,哪位有,能否给我发一份,先谢谢了!email:yjn2005@sohu.com
我们实验室是检测饲料和原料含量的,以前别人介绍用过中国农业科技院分析检测中心研制的氨基酸分析用校核标准品(参比物),名字我忘记了,只记得有代号,1#、2#、3#、4#、5# 这5种。有知道的老师们请帮帮忙!
各位大侠,小弟急需葵二酸的分析方法,哪位大侠请帮帮忙,在此先谢谢了。
各有关单位:依据《中华人民共和国标准化法》《团体标准管理规定》等文件精神,按照《江苏省分析测试协会团体标准管理办法》的有关规定,我会秘书处组织相关专家对南通大学等单位申报的《地表水体中喹诺酮类[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱联用检测法》团体标准进行了立项评审。经专家评审,《地表水体中喹诺酮类[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱联用检测法》团体标准符合立项条件,现批准立项,特此公告。如有单位或个人对该团体标准项目存在异议,请在公告之日起10个工作日内将书面意见反馈至江苏省分析测试协会秘书处。联系人: 周 明电 话:13770810997E-mail:jsfxcsxh@163.com[align=right]江苏省分析测试协会[/align][align=right]2022年9月19日[/align][url=http://file2.foodmate.net/wenku2022/wfx202209220939.zip]附件[/url]:江苏省分析测试关于《地表水体中喹诺酮类[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱联用检测法》团体标准的立项公告.pdf
哪位朋友知道欧盟水产食品中氟喹诺酮标准啊?谢谢!
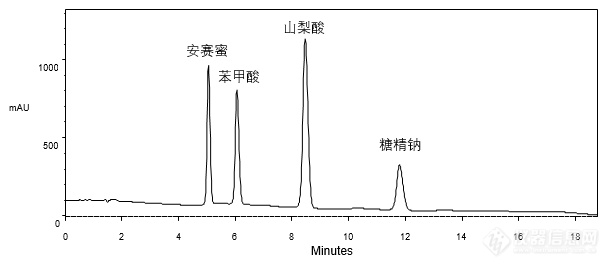
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
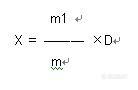
标准简介:GB/T 8381.10-2005 饲料中磺胺喹恶啉的测定高效液相色谱法 本标准规定了以高效液相色谱(HPLC)仪测定饲料中磺胺喹恶啉的方法。本标准适用于配合饲料、浓缩饲料和添加剂预混合饲料中磺胺喹恶啉的测定,最小检测浓度为5.0 mg/kg。磺胺喹恶啉属磺胺类抗生素兼有抗球虫作用,广泛用于养禽业,其口服后吸收迅速,但排泄缓慢,残留在组织器官及鸡蛋中时间长,人食用后,对人体产生耐药性等副作用。世界卫生组织(WHO)规定动物组织、奶的最高残留限量(MRL)值为100μg/kg。日本等国规定肉鸡中不得检出。因此,该药的使用应严格控制用药期及用药量,以保证产品的安全及对外贸易畅通。英国兽药典记载,磺胺喹恶啉纯品为黄色粉末,几乎无味,不溶于水,溶于甲醇、乙醇,易溶于碱性溶液。目前,饲料中磺胺喹恶啉的检测方法美国AOAC标准是采用分光光度法,该法要求含阿散酸及不含阿散酸的饲料采用两种不同的测定方法。而国内饲料目前添加组分较复杂,检测者对饲料情况是未知的,难以确定用哪一个方法;且该方法中需要重氮化试剂做偶合反应,其毒性大,对操作者及环境 都会造成危害。国标GB/T 8381.10中,规定了采用高效液相色谱法测定饲料中磺胺喹恶啉。方法简介如下:1.测定方法1.1原理 用甲醇水溶液提取饲料中的磺胺喹恶啉,离心,过滤,在HPLC仪上分离,紫外检测器240nm处测定。1.2 试剂和溶液以下所用的试剂,除特别注明外均为分析纯试剂;水为蒸馏水,色谱用水符合GB/T6682规定的一级水。1.2.1 磺胺喹恶啉标准品:含磺胺喹恶啉(C14H12N4O2S) 95.0%。1.2.2 甲醇:色谱纯。1.2.3 磷酸盐溶液:取磷酸二氢钾3.40g和磷酸氢二钾5.71g,加水溶解并稀释至1000mL。1.2.4 磺胺喹恶啉标准溶液:准确称取磺胺喹恶啉(4.1)50mg,溶于甲醇并稀释成0.1mg/mL的储备液,置4℃冰箱中避光保存,有效期1个月。临用前,取此储备液用水稀释成适当浓度的标准工作液。1.2.5 提取液:甲醇100mL+水50mL1.3 仪器1.3.1 实验室常用仪器设备1.3.2高效液相色谱仪(配紫外检测器)1.3.3 分析天平:感量为0.0001g和0.001g1.3.4 旋涡振荡器1.3.5 离心机:4000r/min1.3.6针头过滤器:备孔径为0.45μm 微孔滤膜1.4 分析步骤1.4.1 提取 称取5g试样,精确至0.001g,加入提取液(4.5)50mL,旋涡振荡器混匀,超声水浴中提取15min,中间取出摇动1次,然后4000r/min离心5min,静置,取上清液过0.45μm滤膜,供液相色谱测定。1.4.2 标准曲线的制备 准确吸取储备液适量,用水或流动相稀释成浓度分别为0.10、0.50、1.00、2.00、10.0μg/mL的磺胺喹恶啉标准溶液,做出标准曲线。 1.4.3 测定1.4.3.1 色谱条件 色谱柱:C18柱 柱长150mm,柱内径4.6mm,粒度5μm或性能相当者。 流动相:磷酸盐溶液75mL+甲醇25mL,用前过0.45μm滤膜,并超声脱气。 流速:1mL/min。 检测波长:240nm。 进样量:10~20μL。1.4.3.2 定性与定量 根据标准品的保留时间定性,定量由标准曲线或单点校准。1.4.4 结果的计算 每千克试样中含磺胺喹恶啉的质量按下式公式计算: http://ng1.17img.cn/bbsfiles/images/2010/11/201011131559_259214_1620630_3.jpg x——每千克试样中磺胺喹恶啉的质量(mg); m1——色谱峰面积对应的磺胺喹恶啉的质量(μg); D——稀释倍数; m——所称样品的质量(g)。 平行测定结果用算术平均值表示,保留至小数点后1位。
请问乙二胺四乙酸分析纯能直接配制EDTA标准溶液吗?请如果能配制有方法吗,比如配0.1mol/L的,请各位帮忙,我手里现在只有乙二胺四乙酸分析纯

抗生素头孢喹肟的动物体内药动学分析 头孢喹肟是目前唯一一个动物专用第四代头孢类抗生素,具有抗菌谱广,抗菌活性强的特点,适用于非肠道用药;源于基本头孢菌素结构的化学修饰提供了头孢喹肟的两性离子性质。头孢喹肟的这一特性可以促进其迅速跨生物膜渗透作用(包括细菌细胞壁的孔蛋白),从而增强生物利用度,较第二代和第三代头孢菌素抗菌谱更广。它对临床重要细菌的染色体和质粒编码的β-内酰胺酶高度稳定。被用于治疗动物呼吸道的疾病,牛的急性乳腺炎和腐蹄病,小牛败血症,猪子宫炎,乳房炎,无乳综合征,马驹败血病,其也是治疗羊的各类疾病的药物。 材料和方法: 头孢喹肟,、色谱乙腈、色谱甲醇、三氟乙酸(TFA)、去离子水。 岛津高效液相色谱仪、SPD-10AVP UV-VIS检测器268纳米处进行检测、柱温箱40°C、Phenomenex Gemini C18色谱柱 (250 mm ×4.6 mm; 5u m)。 流动相为乙腈和0.1%三氟乙酸水溶液,以0.9 ml/min的流速进行洗脱。 标准溶液配制: 头孢喹肟的储备溶液通过直接称量干燥后的标准物质溶解于水中,浓度1mg/ ml,并将该溶液保存于-70℃。头孢喹肟标准溶液通过加入空白血浆配制成溶度为0, 0.02, 0.04, 0.10, 0.40, 1, 2, 4, 10,和 12 ug/ml的溶液。 样品制备:200ul血浆中加入1.5毫升微量离心管中,加入等体积的甲醇使蛋白质沉淀,离心(4000转/min)10分钟后,300ul的上清离心液移入新鲜小瓶中,加入150ul去离子水混合,再吸取50ul上清液进样分析。 液相方法的验证: 选择性-选择性通过分析空白血浆、加入头孢喹肟的血浆、从羊服用头孢喹肟药代动力学研究中获得的血浆样品进行评价,无内源性化合物对目标化合物的干扰。 线性标准曲线-本方法的线性通过在0.02-12ug/ml的范围内的校正曲线评价。 灵敏度-通过进样 0.01ug/ml-0.1 ug/ml评价其信噪比。 精密度和准确度-头孢喹肟样品分低中高浓度分别进样分析,每个浓度分别进样六次日间、日内测定。 回收率-为了计算头孢喹肟的绝对回收率分别加入 0.4ug/ml, 2ug/ml, 和10ug/ml浓度样品,每个浓度分别重复6次进行加入和提取。 稳定性-标准
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7691601521 二类残留溶剂-1,4-二氧;六环 Residual Solvent Class 2 - 1,4-Dioxane 对照品/标准品1601500 二类残留溶剂-N,N-二甲基酰胺 Residual Solvent Class 2 - N,N-Dimethylformamide 对照品/标准品1601485 二类残留溶剂-N,N-二甲基乙酰胺 Residual Solvent Class 2 - N,N-Dimethylacetamide 对照品/标准品1601463 二类残留溶剂-1,2-二甲氧基乙烷 Residual Solvent Class 2 - 1,2-Dimethoxyethane 对照品/标准品1601441 二类残留溶剂-二氯甲烷 Residual Solvent Class 2 -Methylene Chloride 对照品/标准品1601420 二类残留溶剂- 1,2- 二氯乙烯 Residual Solvent Class 2 - 1,2-Dichloroethene 对照品/标准品1601408 二类残留溶剂-环己烷 Residual Solvent Class 2 - Cyclohexane 对照品/标准品1601383 二类残留溶剂-氯仿 Residual Solvent Class 2 -Chloroform 对照品/标准品1601361 二类残留溶剂-氯苯 Residual Solvent Class 2 - Chlorobenzene 对照品/标准品1601340 二类残留溶剂-乙腈 Residual Solvent Class 2 -Acetonitrile 对照品/标准品1601306 二类残留溶剂-混合物 C Residual Solvent Class 2 - Mixture C 对照品/标准品1601292 二类残留溶剂-混合物 B Residual Solvents Class 2 - Mixture B 对照品/标准品1601281 二类残留溶剂-混合物 A Residual Solvents Class 2 Mixture A 对照品/标准品1601226 一类残留溶剂- 1,1,1- 三氯乙烷 Residual Solvent Class 1 -1,1,1 对照品/标准品1601204 一类残留溶剂- 1,1- 二氯乙烯 Residual Solvent Class 1 -1,1-Dichlo 对照品/标准品1601180 一类残留溶剂- 1,2- 二氯乙烷 Residual Solvent Class 1 -1,2-Dichlo 对照品/标准品1601168 一类残留溶剂-四氯化碳 Residual Solvent Class 1 -Carbon Tetrachloride 对照品/标准品1601146 一类残留溶剂-甲苯 Residual Solvent Class 1- Benzene 对照品/标准品1601102 一类残留溶剂混合物 Residual Solvents Mixture Class 对照品/标准品1601000 利血平 Reserpine 对照品/标准品1600846 瑞格列奈杂质C Repaglinide Related Compound C 对照品/标准品1600835 瑞格列奈杂质B Repaglinide Related Compound B 对照品/标准品1600824 瑞格列奈杂质A Repaglinide Related Compound A 对照品/标准品1600813 瑞格列奈 Repaglinide 对照品/标准品1600121 瑞鲍迪甙 A Rebaudioside A 对照品/标准品1599500 红车轴草提取粉 Powdered Red Clover Extract 对照品/标准品1599000 萝芙碱 Rauwolfia Serpentina 对照品/标准品1598802 树莓酒 Raspberry Alcohol 对照品/标准品1598700 雷尼替丁杂质C Ranitidine Related Compound C 对照品/标准品1598609 雷尼替丁杂质B Ranitidine Related Compound B 对照品/标准品1598507 雷尼替丁杂质A Ranitidine Related Compound A 对照品/标准品1598450 雷尼替丁分离度用混合物 Ranitidine Resolution Mixture 对照品/标准品1598405 盐酸雷尼替丁 Ranitidine Hydrochloride 对照品/标准品1598347 雷米普利杂质D (二酮哌嗪雷米普利)Ramipril Related Compound D 对照品/标准品1598338 雷米普利杂质C Ramipril Related Compound C 对照品/标准品1598323 雷米普利杂质B Ramipril Related Compound B 对照品/标准品1598314 雷米普利杂质A Ramipril Related Compound A 对照品/标准品1598303 雷米普利 Ramipril 对照品/标准品1598201 盐酸雷洛昔芬 Raloxifene Hydrochloride 对照品/标准品1598008 3- 奎宁环基 3-Quinuclidinyl Benzilate 对照品/标准品1597504 奎宁酮 Quininone 对照品/标准品1597005 硫酸奎宁 Quinine Sulfate 对照品/标准品1596807 二水合盐酸奎宁 Quinine Hydrochloride Dihydrate 对照品/标准品1595509 硫酸奎尼丁 Quinidine Sulfate 对照品/标准品1595000 葡萄糖酸奎尼丁 Quinidine Gluconate 对照品/标准品1594506 金鸡纳酸 Quinic Acid 对照品/标准品1594007 喹乙宗 Quinethazone 对照品/标准品1593423 喹那普利杂质 B Quinapril Related Compound B 对照品/标准品1593412 喹那普利杂质 A Quinapril Related Compound A 对照品/标准品1593401 盐酸喹那普利 Quinapril Hydrochloride 对照品/标准品1593004 盐酸米帕林 Quinacrine Hydrochloride 对照品/标准品1592409 槲皮素 Quercetin 对照品/标准品1592227 夸西泮杂质 A Quazepam Related Compound A 对照品/标准品1592205 夸西泮CIV Quazepam CIV 对照品/标准品1592001 恩波吡维铵 Pyrvinium Pamoate 对照品/标准品1589109 丙酮酸 Pyruvic Acid 对照品/标准品1589007 乙胺嘧啶 Pyrimethamine 对照品/标准品1588004 马来酸吡拉明 Pyrilamine Maleate 对照品/标准品
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心

蛮好的东西,可惜要过期了。近期有做这些实验的老师,赶快联系我司业务员吧,亏本销售,买了就是赚了!产品质量保证,您大可放心。详细信息: 1. C17396150 四环素盐酸盐 标准品 0.25g Tetracycline hydrochloride CAS 有效期 2012/05 价格180RMB2. C10745000 甲基溴硫磷 标准品 0.1g Bromophos-methyl CAS 有效期 2012/05 价格240RMB贴几张图看看,很好的东西,平时这个价格全中国那也买不到呀。http://ng1.17img.cn/bbsfiles/images/2012/02/201202241905_350946_2378824_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/02/201202241907_350947_2378824_3.jpg
跪求有没有人有“硫酸银、碳酸银”分析标准啊?
食品分析中标准物的管理及其标准溶液的校正一、意义食品分析标准物质是分析方法质控的核心,是定性和定量的依据。标准物质以一定纯度和浓度配制的溶液称标准溶液,其稳定性受其自身降解、化学转化、溶质和溶剂挥发等内在因素的影响,又受其存放条件如温度、湿度、光线照射、存放时间、存放容器及其配制技巧等外界条件的影响。由于影响标准物及其标准液的因素较多,如果条件控制不当或管理不严密,标准物质浓度易发生变化,这是食品分析中难以进行质量控制的主要原因。以上原因也是实验室食品分析测定产生误差的最主要因素,要减小这些产生误差的主要因素,就要特别注重标准物管理,以及进行标准溶液的稳定性观察和校正工作,也是实验室质量控制关键工作之一。当标准溶液发生变化时就要重新配制,并找出变化原因,为分析工作积累经验,并可写入方法注释中。在食品分析质量控制中,准确度和精密度的提高,是以标准溶液稳定性和准确性为前提的,因此对于标准溶液稳定性和准确性的关键技术问题,是质量控制的核心问题。二、标准物质的管理 1、容量分析的基准物是标定其它标准溶液的基准,应购买基准试剂,它可以保证其纯度。另一个因素是水份的影响,用前应充分干燥和恒重,配制时量器要校正。溶剂要纯化,使用的容器要充分洗涤。最终目的都是防止基准物的化合损失。2、用于分光光度分析的标准物,分有机的和无机的标准物,无机标准稳定性好,但使用液浓度低时,极易被容器吸附,并与容器中离子进行交换,因此决定了其稀溶液使用时间短。玻璃容器在碱性介质中易溶出,塑料容器在成型时加入助剂时也含有不同金属杂质,容易溶出如Zn、Ca等金属离子。有机标准最好不放在塑料制容器中,因塑料在成型时加入的有成份比较复杂的助剂和增塑剂。标准使用液应现用现配为最佳。三、标准物存放使用1、无机的标准物要求在干燥并无化学干扰物的条件下存放,选择合适干燥剂如硅胶和分子筛。有机标准物最好分装封入安培瓶中低温避光保存。固体的多环芳烃可配制成溶液,再分装在安瓿瓶中保留溶剂封存,也可把溶剂挥掉后干燥封存,使用时再定容,后一种方法更为稳妥些。配制好一批标准溶液,再一支一支使用也是很方便的。如果液体的标准物特别是几种标准的混合物用于色谱分析同系物如醇类物质,可同时配制一批分装安瓿瓶低温存放,再一支一支使用,能避免溶剂挥发体积变化产生的误差。这种做法更适于实验室间的标准分发和校正工作。最难办的是气体,标准如氯乙烯、氟里昂,最好是钢瓶中存放,或配制钢瓶标准气。这些条件不具备时也可以选择高沸点溶剂,密封溶解这些气体,称量溶质重量,一次性使用。从这一事实出发,气体,测定误差可以稍加放宽,因为标准自身稳定性差。2、用于色谱分析的标准物要求色谱纯,其配剂溶液剂也要求色谱纯,准确配制前要在色谱上进行检查。特别是几种标准进行混合更应慎重,每种要严格检查否则给定性定量带来很多麻烦。如果纯度不够时可以纯化,再结晶或用制备色谱制备。勉强使用是无益的。四、标准溶液的校正1、从安瓿瓶分装标准溶液无论是有机的或是无机的用于校正是很方便的。如原子吸收测定金属,从安瓿瓶中取一定量配制浓度系列,再封存。每隔一段时间(1~2周)再用原溶液配制同样浓度系列,严格控制仪器条件来比较二次标准曲线的斜率,斜率下降时表明有损失。2、相同浓度同时配制的标准的几支安瓿瓶,先用打开的一支标准的测定值与间隔一段时间后打开的另一支标准的测定值进行比较,以此类推最后在一段时间内几支同时测定,其变异程度就是标准在这段时间稳定性变化程度。3、几个实验室用同一标准物分别配制相同浓度标准液,各自进行标准曲线的测定,再按规定交换该标准液再进行测定,比较测定结果差异来观察同一标准的时间和空间变异。如果标准液稳定,配制不准确的实验室很容易查出。配制都准确时,标准液若不稳定时,会使各实验室的测得值都偏低。4、同种标准物来源不同,也应采用分别配制交叉测定的办法来检查标准的纯度及配制是否准确。在食品分析中无论用何种手段分析样品,所使用的标准物应作统一的或确切的规定。例如:过硫酸铵测锰,用MnSO4·H2O作为标准使用,到底硫酸锰需要不需要烘烤呢?对于这个问题,在一部份的教科书中有规定烘烤的,也有不烘烤的。按照MnSO4·H2O的性质遇到空气可能吸潮或风化,如果直接称重计算Mn量,就有可能出现误差。用烘烤称重测得水分所含的量比理论值高1.6%,有同一硫酸锰配制锰标准溶液测一合成水样,使用烘烤后配制的锰标准溶液,测得的Mn含量为0.205mg/L,未烘烤过的则高达约2.3%,从中说明硫酸锰在配制标准溶液时应经过烘烤,使标准一致。
3-硫酸甘氨鹅脱氧胆酸 标准品,有可以联系我renzhichu525@163.com.
成分分析中硫酸法测定棉和聚酯纤维的含量时,标准要求1克样品200ML硫酸,这量比较大,大家有用100ML硫酸做过实验吗?
制作龙葵素标准曲线 没有用空白调零 试验组中龙葵素吸光度可以用试验组减去空白组的吸光度算么
求助:原乙酸三甲酯分析方法或分析标准
最近发现用ICP分析Sb元素,弱酸介质下GBW配置的标准曲线分析的其他品牌的标准溶液回收率偏低。实验室用的仪器是瓦里安 ICP-720,标准曲线是使用国家计量院的GBW(E)080545锑单元素溶液标准物质,浓度100ug/mL,基体是5%HCl。作为交叉验证的CK是使用国家钢铁材料测试中心钢铁研究总院的GSB G 62043-90锑标准溶液,浓度500ug/mL,基体是25%硫酸。或者是ACCU的ICP-02N-1,浓度1000ug/mL,基体是2~5%HNO3。我发现使用弱酸基体(若0.07mol/L HCl或者5%HNO3)配置的标准曲线(使用计量院的Sb标准溶液配置),分析相同基体的CK(由另外品牌的Sb标准溶液配置),测量第一个CK时,Sb回收率只有70-85%(酸度越低,回收率越差),继续测量,回收率会慢慢增大。可是即使连续测量(不拔出进样管)10次以上,Sb的回收率也只有93-94%。而测量由计量院的Sb标准溶液配置的同样基体的Sb溶液,回收率却没有问题;若是使用浓酸基体(35%HNO3),两个品牌的标准溶液的回收率却又没有问题。我知道Sb会有残留,可是分析每一只样品(包括标准曲线的点),我都会快泵进样十几秒再分析溶液的,就算有残留,没道理厉害到分析了10多样品还有残留。母溶液的基体可能有影响,可是同一支溶液的其他元素的回收率都OK啊最近有支PT样(5%硝酸基体,什么品牌的母溶液配出来的就不知道了),就是因为这个原因,Sb的读数偏低了。现在要整改,可是什么原因都不知道……现在只好到论坛来求助各位老大了,希望各位能给点意见。先谢谢了。
[size=4] 喹诺酮为一类具有4一喹诺酮环结构的药物。第一代药物萘啶酸(1962),第二代药物吡哌酸和氟甲喹(1974),抗菌作用较弱,国内较少使用。第j代为氟喹诺酮类(具有6一氟一 7一哌嗪一4一诺酮环结构)。喹诺酮类药物结构相似,取代位点较多,抗菌谱较广,活性高,从其结构一活性关系上探索开发新品种己成为喹诺酮类药物的研究热点,因而发展迅速, [/size][size=4] 尤以人药领域的喹诺酮类药物发展为最陕。最近几年又推出了数十种之多的新品种,其中有些还未命名,只给出了试验编号。 [/size][size=4] 喹诺酮类药物广泛地用于畜禽的细菌、霉形体病防治,已投人使用或即将进人兽医领域的药物有10多种,主要有两类,一类从人医用移植转化而来,如诺氟沙星、环丙沙星、氧氟沙星、培氟沙星、洛美沙星等。另一类是动物专用品种,己批准上市的兽医专用喹诺酮类药物有恩诺沙星(德国拜耳公司)、沙拉沙星(美国雅培公司)、单诺沙星(美国辉瑞公司)、二氟沙星(美国雅培公司)、倍诺沙星(日本武田制药),奥比沙星(日本大日本制药)和麻保沙星(瑞士罗氏公司),其中,后三种在我国还未见上市。 [/size][size=4] 诺酮类药物与细菌DNA复制所需的DNA一~rase的亚基A(Subunit)结合而抑制DNA复制化,此外由于细菌细胞具有强烈的穿透力,故具有强大的杀菌作用。这些药物的抗菌作用与疗效可与第j代头孢菌素媲美,已成为兽用抗菌药物中最活跃的研究领域之一,随之而来的这类药物的分析分析显得十分重要,相关文章也比较多,但大多数文章,例如彭六保等从四代喹诺酮类药物分类综述了该类药物的分析进展,其针对性不强;还有一些文章,如王玉忠、张加玲等从各种分析方法进行综述,与上面存在同样的问题。所以本文仅对兽医常用的九种喹诺酮类药物分析进展综述,以期对兽药临床及生产具有一定的帮助。现将常用的分析方法方法介绍如下。 [/size]
手上有棕榈酸甲酯和硬脂酸甲酯的标准品各50mg,请问该怎么处理进气相会比较好?另外,关于脂肪酸的定量分析是用内标法比较好吗?外标法行吗?那这样的话稀释浓度梯度该为多少比较合适?拜托各位老师教教我..先.谢谢了!!
谁有新的高纯碳酸锂分析标准?急用,谢谢
化验分析硅石标准品,怎样测铝的含量,标准品如何处理?
刚刚看到国标GB/T 22110-2008 食品中反式脂肪酸的测定-气相色谱法 中用到反-9、12、15十八碳三烯酸甲酯标准品(trans-9,trans-12,trans-15-Octadecatrienoic acid methyl ester),有谁买过,哪个品牌可以提供?谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP