在薄层色谱定性时,对照品、供试品和对照药材在相同的问题显相同颜色的斑点即可。对照品和对照药材定性有何区别?为什么两个要同时做呢?

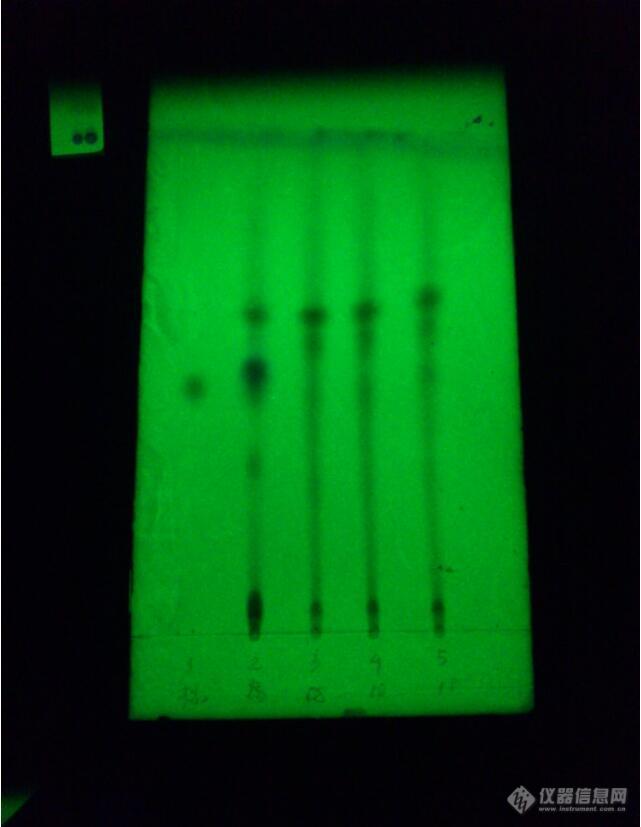
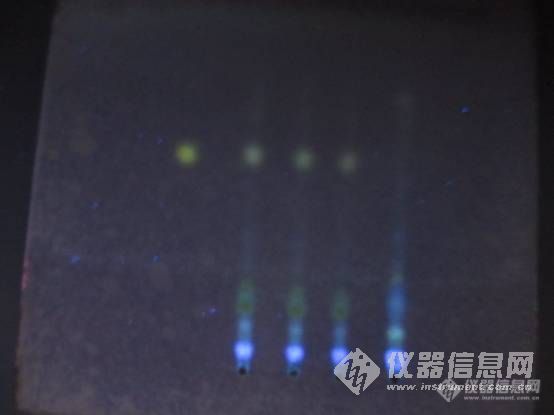
中药制剂中木瓜的薄层鉴别:供试品与对照药材处相应的点颜色不同是为什么?狗脊的薄层鉴别也是一样,提取时处理方法是一样的图1前三供试品(绿色),中二木瓜对照药材(蓝色),后二阴性对照图2前二供试品(亮的点蓝色),中二狗脊对照药材(绿色),后二阴性对照不知道该怎么办,诚请各位大师解答,谢谢[img=,690,459]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329352099_2969_1816508_3.jpg!w690x459.jpg[/img][img=,380,362]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329533546_2463_1816508_3.jpg!w380x362.jpg[/img]
按照药典规定的,展开剂:氯仿-醋酸乙酯-丙酮-甲酸=6:2.5:2.5:0.21为原儿茶酸对照品2药材对照品3、4、5均为制成的中药制剂供试品。我已经增大浓度试过了,色谱图无差异[img=,641,827]https://ng1.17img.cn/bbsfiles/images/2019/08/201908140947125804_8826_1791505_3.jpg!w641x827.jpg[/img]
研究中药复方制剂中各味药材的鉴别药典中鉴别乌梅药材,选择“熊果酸”为对照乌梅含有熊果酸,大枣也含有熊果酸,请问那我做制剂中乌梅药材鉴别时,是不是就不能以熊果酸作对照了,因为怕大枣会有干扰?这种情况,乌梅该选择什么组分作为对照?实验结果显示,乌梅阴性并没有干扰,我能以“熊果酸”作为对照鉴别乌梅吗,虽然明知大枣也含熊果酸。乌梅阴性没有干扰,会不会是点样量少的问题?http://ng1.17img.cn/bbsfiles/images/2012/11/201211010017_400582_1872149_3.jpg1. 熊果酸;2-4. 样品;5. 阴性
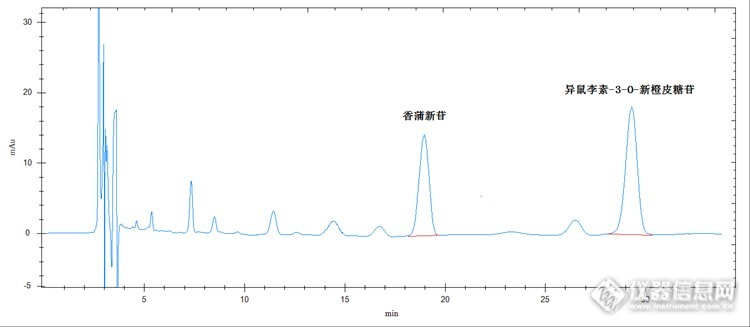
好药材不怕检,蒲黄药材接着检 蒲黄药材为香蒲科植物狭叶香蒲、宽叶香蒲、东方香蒲和长苞香的花粉,具有止血,化瘀,通淋功效。用于吐血,衄血,咯血,崩漏,外伤出血,经闭痛经,脘腹刺痛,跌扑肿痛,血淋涩痛效果较好。实验部分原理 取适量该药材,加甲醇溶解,加热回流或超声波提取,经进样系统进样,色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量计算。仪器及试剂 仪器:高效液相色谱仪(紫外检测器),柱温箱,超声波清洗仪,溶剂过滤器,针筒式过滤器,加热回流装置,电子天平 试剂:甲醇(色谱纯),超纯水样品制备 对照品溶液的制备:精密称取异鼠李素-3-O-新橙皮糖苷、香蒲新苷对照品适量,加甲醇配制成浓度均为50μg/ml的对照品溶液,备用。 供试品溶液的制备:精密称取本品约0.5g,置具塞锥形瓶中,精密加入50ml甲醇后,称定重量并记录,冷浸12小时后加热回流1小时(或超声波超声30min),放冷,再次称定重量,用甲醇补足减少的重量,摇匀,滤过,待测。色谱条件检测器:紫外检测器色谱柱:C18,4.6 X 250mm,5μm流动相:乙腈:0.05%磷酸溶液=15:85(V:V)检测波长:254nm进样量20μl柱温:室温对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192152_519002_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519003_2498430_3.png 从以上色谱图我们可以看出样品出峰时间很晚,峰形也较差。下面我们换用一根耐酸性色谱柱,效果我们请看色谱图。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519004_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519005_2498430_3.png 换了这根色谱柱,色谱图的峰形好了很多,出峰时间也明显有所提前,但保留时间还是有点晚。下面我们又把色谱柱温度调整了一下,调到了40℃,效果接着往下看。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410192153_519006_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/10/201410231859_519702_2498430_3.png 当然保留时间还可以再缩短缩短(通过增加流动相中甲醇含量,或提高高压泵流速,或换用更高效更短的色谱柱),但供试品中异鼠李素-3-O-新橙皮糖苷的附近有一个干扰物,为了保证分离度,这个分析时间已经比较合理,不要再缩短了。 检测蒲黄药材的这个方法到现在已经很完美了。但有几点事项需要注意。1.样品若采用超声波超声提取,为了保证提取效果有时得增加超声时间或超声波水域温度。2.检测这个样品最好要选择一款效果好的色谱柱,如耐酸性的色谱柱。3.为了缩短检测时间我们可以升高色谱柱的温度,对于这个样品效果就挺好。当然适当增加流动相中甲醇含量或增加高压泵流速,或换用更高效更短的色谱柱也能达到比较理想的效果。

请各位老师帮忙看看,不知荆芥对照药材(右边一个为对照药材,左边三个为制剂样品——含荆芥)本身就这样,还是我做的不好,斑点不清晰。多谢![img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907171127582678_7811_1825519_3.jpg!w690x517.jpg[/img]

黄连上清片高温灭菌后,黄连对照药材薄层鉴别的点变红是怎么回事?[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2019/07/201907101138123673_5943_1782490_3.jpg!w690x388.jpg[/img]
网上“关于明确贵重药材品种的通知”中的药材有以下两个版本“贵重药材”的品种规定如下:麝香、牛黄、 人参、三七、黄连、贝母、鹿茸、虫草、天麻、珍珠、虎骨、豹骨、熊胆、杜仲、厚朴、全蝎、肉桂、沉香、芋肉、蟾酥、银花、巴戟、阿胶、犀角、广角、羚羊角、乳香、没药、血竭、砂仁、檀香、公丁香、西红花。另一个版本是 麝香、牛黄、人参、三七、黄连、贝母、鹿茸、虫草、天麻、珍珠、虎骨、豹骨、熊胆、枸杞、杜仲、厚朴、全蝎、肉桂、沉香、芋肉、竭酥、艮花、马戟、阿胶、犀角、广角、羚羊角、乳香、没药、血竭、砂仁、松香、公丁香、西红花。不知道哪个是对的呢?还有,这个是1981年发的通知,这么多年来有更新过吗?欢迎大家讨论,多谢大家的讨论~~

蒲公英药材高效液相色谱法检测 蒲公英大家应该都不陌生,它春天出土生长,很快开花结果。很多地方它都能生长,山间春林中,田野耕地上,道路旁,小河边,庭院中,沙滩上我们都常会看到它漂亮多瓣的小黄花,褶皱细长的小绿叶,毛茸茸随风飘扬的种子团。它为菊科植物,所以它开的小黄花很像菊花,很漂亮。它的果实为长椭圆形瘦果,上面长有白色冠茸毛,可以随风飘扬,繁衍后代,繁衍能力很强。 蒲公英气微,味微苦。具有清热解毒,消肿散结,利尿通淋等药物功效。可用于治疗疔疮肿毒,乳痈,目赤,咽痛,肺痈,肠痈,湿热黄疸,热淋涩痛等多种病症。 蒲公英中含有多种药物成分和营养成分,其中含量较低的咖啡酸(C9H8O4)具有很好药物疗效,下面我们就采用高效液相色谱法来检测药材蒲公英中咖啡酸含量。实验部分【原理】 精密称取适量蒲公英样品经5%甲酸的甲醇溶液溶解,超声提取,离心、过滤等操作后注入高效液相色谱系统,C18色谱柱分离,紫外检测器检测,外标法(保留时间定性,峰面积定量)计算,得出咖啡酸成分含量。【仪器及试剂】 仪器:高效液相色谱仪(紫外检测器+等度泵+柱温箱+自动进样器+在线脱气机等),超声波清洗仪,溶剂过滤器,电子天平(0.0001),离心机(转速不低于4000r/min)等。 试剂:甲醇(色谱纯),甲酸(分析纯),磷酸(分析纯),磷酸二氢钠(分析纯),超纯水等。【样品制备】 对照品溶液制备:准确称取咖啡酸对照品2mg于50ml容量瓶中,加甲醇溶解、定容,制成40μg/ml咖啡酸对照品溶液,备用。 供试品溶液制备:取蒲公英药材适量,准确称取粗粉1g,置于50ml具塞锥形瓶中,精密加入5%甲酸的甲醇溶液10ml,密塞,摇匀,称定重量,超声处理约30分钟后,取出,放冷,再次称定重量,用5%甲酸的甲醇溶液补足减少的重量,摇匀,离心机离心后,取上清液,0.45um有机相微膜滤过,存储于棕色容量瓶中,待测。【色谱条件】检测器:紫外检测器检测波长:323nm色谱柱:Promosil C18( 250 mm X4.6mm,5μm )色谱柱流动相:甲醇:磷酸盐缓冲液(准确称取磷酸二氢钠1.56g于1000ml容量瓶中,加水溶解定容,用1%磷酸溶液调节pH值至3.9左右)=25:75(V:V)流速:1.0ml/min柱温:40℃进样量:10μl【色谱图】对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/09/201509271303_568091_2536753_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/09/201509271303_568092_2536753_3.png 【结果】 http://ng1.17img.cn/bbsfiles/images/2015/09/201509271303_568093_2536753_3.png 经以上公式计算该蒲公英药材中咖啡酸含量为0.025%,含量较高,高于药典限量要求(药典中要求该含量不得少于0.020%)。【结论】 经重复性、线性、加标回收率等实验验证,该实验方法都具有良好的验证数据。 该检测方法具有检测时间短,分离度好,操作简练,实验数据准确、可靠的优点,适合该样品检测,是该样品检测较为理想的方法。
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体](1)本品粉末棕褐色。果皮石细胞大多成群,黄色,方形或卵圆形,直径7~38[/font][font=宋体]μ[/font][font=宋体]m[/font][font=宋体],壁厚,孔沟明显。种皮石细胞类长方形或不规则形,壁薄,具纹孔。纤维长梭形,直径16~42[/font][font=宋体]μ[/font][font=宋体]m[/font][font=宋体],胞腔较大,壁孔明显。可见梯纹导管和螺纹导管。薄壁细胞不规则形,具纹孔。[/font] [font=宋体](2)取本品粉末1g,加水50ml,超声处理30分钟,滤过,取滤液20ml,加正丁醇振摇提取2次,每次20ml,合并正丁醇液,减压蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取罗汉果对照药材1g,同法制成对照药材溶液。再取罗汉果皂苷V对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各5[/font][font=宋体]μ[/font][font=宋体]l[/font][font=宋体],分别点于同一硅胶G薄层板上,以正丁醇-乙醇-水(8:2:3)为展开剂,展开,取出,晾干,喷以2%香草醛的10%硫酸乙醇溶液,加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font] [b][font=宋体][/font][/b] [font=宋体][/font] [font=宋体][/font] [font=宋体][b]水分[/b] 不得过15.0%(通则0832第二法)。 [/font] [b][font=宋体]总灰分[/font][/b][font=宋体] 不得过5.0%(通则2302)。[/font] [b][font=宋体]【[color=var(--weui-LINK)]浸出物[i][/i][/color]】[/font][/b][font=宋体] 照水溶性浸出物测定法(通则2201)项下的热浸法测定,不得少于30.0%。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 照高效液相色谱法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(23:77)为[color=var(--weui-LINK)]流动相[i][/i][/color];检测波长为203nm。理论板数按罗汉果皂苷V峰计算应不低于3000。[/font] [b][font=宋体]对照品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取罗汉果皂苷V对照品适量,精密称定,加流动相制成每1ml含0.2mg的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取本品粉末(过四号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,加热回流2小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过。精密量取续滤液20ml,回收溶剂至干,加水10ml溶解,通过大孔吸附树脂柱AB-8(内径为1cm,柱高为10cm),以水100ml洗脱,弃去水液,再用20%乙醇100ml洗脱,弃去洗脱液,继用稀乙醇100ml洗脱,收集洗脱液,回收溶剂至干,残渣加流动相溶解,转移至10ml量瓶中,加流动相至刻度,摇匀,即得。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]分别精密吸取对照品溶液与供试品溶液各10[/font][font=宋体]μ[/font][font=宋体]l[/font][font=宋体],注入液相色谱仪,测定,即得。[/font] [font=宋体]本品按干燥品计算,含罗汉果皂苷V (C[sub]60[/sub]H[sub]102[/sub]O[sub]29[/sub])不得少于0.50%。[/font] [font=宋体] [/font]
树皮类药材的采收通常在春夏之交、植物生长旺盛期、树液流动时应尽快采剥。此时,树皮类汁液充足,形成层生长最活跃,皮部与木质部最容易分离,如杜仲、黄柏、厚朴、肉桂等树皮。树皮类药材的采收:通常在春夏之交、植物生长旺盛期、树液流动时应尽快采剥。此时,树皮类汁液充足,形成层生长最活跃,皮部与木质部最容易分离,如杜仲、黄柏、厚朴、肉桂等树皮。其采收方法:一般剥取环状块或采取“剥皮再生法”进行采收。花类药材的采收:这类药材采摘季节性很强,如辛夷花、款冬花、金银花等要采摘未开放的花蕾供药用;绿梅花等要采摘即将开放的花朵入药;菊花、凌霄花、红花、西红花等要采摘盛开的花或花柱供药用。采收方法:选择晴天分期分批采摘,采摘后避免挤压,并注意遮阳,防日晒变色。全草类药材的采收:通常在枝叶生长茂盛、初花时收割,如荆芥、藿香、穿心莲、益母草、半边莲等。但有些应在开花前采收,如佩兰、青蒿等;也有些是采集嫩苗,如春柴胡等;而马鞭草要在花开后采。极少要连根挖出入药,如北细辛、紫花地丁等。采收方法:割取或挖取。叶类药材的采收:一般在植物的叶片生长旺盛、叶色浓绿,花蕾开放前采收,如青叶、紫苏叶、艾叶等品。植物一旦开花结果,叶肉内储藏的营养物质就向花、果转移,从而降低叶类药材的质量。也有极少数叶类药材宜在秋后经霜打后采摘,如桑叶、银杏叶等,而枇杷叶则要在落叶后采。采收方法:摘取、割取或拾取。根及根茎类药材的采收:当植物正在生长发育时,会消耗根部储藏的养分,因此一般在植物休眠期,即秋冬季落叶后至翌年早春萌发前采收根及根茎类药材,如黄芪、党参、丹参、桔梗、丹皮、地骨皮、前胡等。此时地下根和根茎储藏的营养物质和有效成分含量最高。少数药材如白芷、当归、川芎等应在生长期采收。采收方法:选雨后的晴天或阴天,在土壤较湿润时用锄头或特制的工具挖取。采挖时注意保持根皮完整,避免损伤而降低药材质量。根皮类药材的采收:采收时期同根茎类。先将根部从土中挖出,然后进行砸打或搓揉使皮肉与木心分离,如五加皮、远志肉等根皮。果实类药材的采收:多数果实类药材在果实完全成熟时采收,如瓜蒌、黄栀子、薏苡仁、花椒、八角等;也有些要求果实成熟经霜打后再采,如山茱萸霜后变红、川楝子霜打变黄时才采收;还有些应在果实未成熟时采收,如青皮、枳实、桔红等。果实成熟期不一致的药材,如山楂等,要随熟随采,过早采收肉薄产量低,过期采收肉松泡,质量差。多汁浆果,如枸杞子、山茱萸等,采摘后应避免挤压和翻动。采收方法:摘取或剪取。同一果序上的果实成熟期一致的,如女贞子、五味子等,可将整果序剪取,放置若干天后摘取果实。种子药材的采收:多数种子类药材要在果实充分成熟、籽粒饱满时采收,如牵牛子、决明子、补骨脂、续断子等。一些蒴果类的种子,若待果实完全成熟,则蒴果开裂,种子散失,难以收集,须稍提早采收,如急性子、牵牛子、豆蔻等。对种子成熟期不一致而且成熟即脱落的药材,如补骨脂等,应随熟随采。干果类一般在干燥后取出种子,蒴果通常敲打后收集。肉质果,若果肉亦作药用的,可先剥取果肉,留下种子或果核,如瓜蒌子等;有些果肉不能作药用的则取出种仁。
[font=Arial, 'Microsoft Yahei'][color=#333333]目的[/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333] 建立固公果根药材的超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联四极杆-静电场轨道阱质谱(UPLC-Q-Exactive Orbitrap-MS)指纹图谱,并研究其与自由基清除活性的谱效关系。 [/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333]方法[/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333] 采用UPLC-Q-Exactive Orbitrap-MS建立固公果根的指纹图谱,评价指纹图谱相似度,结合热图分析,对其中的19个共有峰进行指认 使用1,1-二苯基-2-三硝基苯肼(DPPH)法评价固公果根的抗氧化活性,结合灰色关联度分析(grey relational analysis,GRA)研究谱效关系。 [/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333]结果[/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333] 建立了固公果根的UPLC-Q-Exactive Orbitrap-MS指纹图谱,选择峰面积1%的19个色谱共有峰,通过对照品比对法、数据库、参考文献等方法鉴定前述色谱峰。19个色谱峰的热图分析和DPPH抗氧化灰色关联度结果显示,面积颜色变动较大峰为2、4、8、14、15、17,谱效关系表明11、18、17、19、8、6、16、12、1、5、7、15号峰是与抗氧化活性关联较大的正相关峰。各取排名前五相交得以下9个化学成分作为质量评价指标:峰2(儿茶素)、峰4(表儿茶素)、峰8[(+)-喹色亭酚-(4β,8)-表儿茶素]、峰11(鞣花酸)、峰14(野蔷薇苷)、峰15(刺梨苷)、峰17(委陵菜酸)、峰18(蔷薇酸)和峰19(皂皮酸)。 [/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333]结论[/color][/font][font=Arial, 'Microsoft Yahei'][color=#333333] 固公果根抗氧化活性是酚类和三萜皂苷类等多成分联合效应的结果,为药材质量控制提供更全面的参考。[/color][/font]
白芷的荧光鉴别白芷是与肉桂、红花、川乌、草乌、荆芥、防风、干姜、金银花、当归、三棱、莪术混提,用水煎煮提取2次,每次2小时,成品中有白芷的薄层鉴别,与白芷对照药材在紫外下观察荧光。我已经做了多批,都看不到荧光斑点,很困惑,请有经验的老师帮忙指导。
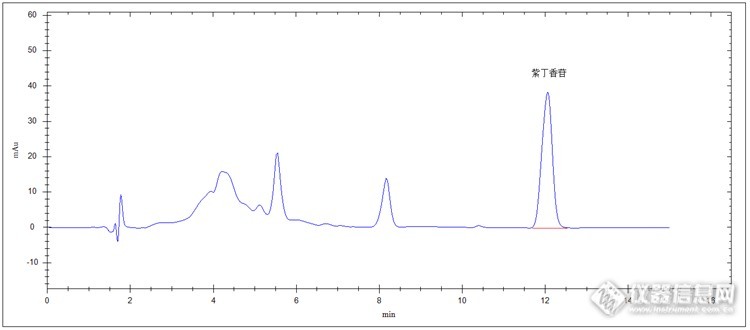
中药材刺五加检测 中药材刺五加为五加科植物刺五加的干燥根和根茎。可在春、秋二季采收,清水洗净,晒至干燥。 刺五加具有益气健脾、补肾安神、强身健体、补肾安神、延年益寿、安神醒脑功效。可用于治疗脾肺气虚、体虚乏力、食欲不振、腰膝酸软、肺肾两虚、久咳虚喘、腰膝酸痛、失眠多梦。另外对脑动脉硬化、脑血栓、脑栓塞、冠心病、神经衰弱、更年期等病症也有很好疗效。 刺五加药物功效很多,是很多药品生产的原材料,所以它中重要药物成分含量需要严格控制。下面我们介绍高效液相色谱法检测中药材刺五加中紫丁香苷含量实验。原理 取适量该粉末药材,加甲醇溶解,超声波超声提取,进样器进入高效液相色谱系统,由流动相带人C18色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量(外标法)计算。仪器及试剂 仪器:高效液相色谱仪(紫外检测器+高压输液泵+柱温箱+自动进样器+在线脱气机等),超声波清洗仪,溶剂过滤器,电子天平,三号药典筛等。 试剂:甲醇(色谱纯),超纯水等。对照品溶液制备 精密称取紫丁香苷对照品2mg与25ml容量瓶中,加甲醇溶解并至刻度,配制成80μg/ml紫丁香苷对照品溶液,备用。供试品溶液制备 取刺五加药材适量,充分粉碎后过三号筛(药典筛),精密称取过筛后粉末2g于具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声处理大约30分钟,放冷,再次称定重量,用甲醇补足减少的重量,摇匀,0.45um有机相微膜滤过,待测。色谱条件检测器:紫外检测器色谱柱:pGrandsil-STC-C18,4.6 X 250mm,5μm流动相:甲醇-水(25:75)检测波长:265 nm流速:1.0 mL/min柱温:30℃进样量:10ul对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2014/12/201412282227_529738_2498430_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &][size=16px][/size][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体](1)本品种子横切面:假种皮薄壁细胞含淀粉粒。种皮表皮细胞棕色,长方形,壁较厚;下皮细胞1列,含黄色物;油细胞层为1列油细胞,类方形或长方形,切向42~162[/font][font=&]μm[/font][font=宋体],径向48~68[/font][font=&]μm[/font][font=宋体],含黄色油滴;色素层为数列棕色细胞,皱缩。内种皮为1列栅状厚壁细胞,棕红色,内壁与侧壁极厚,胞腔小,内含硅质块。外胚乳细胞含淀粉粒和少数细小草酸钙簇晶及方晶。内胚乳细胞含糊粉粒和淀粉粒。[/font] [font=宋体](2)取(含量测定)项下的挥发油,加乙醇制成每1ml含50[/font][font=&]μl[/font][font=宋体]的溶液,作为供试品溶液。另取桉油精对照品,加乙醇制成每1ml含20[/font][font=&]μl[/font][font=宋体]的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各1[/font][font=&]μl[/font][font=宋体],分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(17:3)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同的蓝色斑点。[/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][font=宋体][/font] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体] 水分[/font][/b][font=宋体] [/font][font=宋体]不得过15.0%(通则0832第四法)。[/font] [b][font=宋体]总灰分 [/font][/b][font=宋体] [/font][font=宋体]不得过8.0%(通则2302)。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 照挥发油测定法(通则2204)测定。[/font] [font=宋体]本品种子团含挥发油不得少于1.4%(ml/g)。[/font] [font=宋体][/font][font=宋体][/font] [color=#93c6bc][size=20px][b]其他[/b][/size][/color][size=16px][color=#e2a4a4]|[/color][/size][b][font=宋体][/font][/b] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体]【炮制】 草果仁[/font][/b][font=宋体] [/font][font=宋体]取草果,照清炒法(通则0213)炒至焦黄色并微鼓起,去壳,取仁。用时捣碎。[/font] [font=宋体]本品呈圆锥状多面体,直径约5mm;表面棕色至红棕色,有的可见外被残留灰白色膜质的假种皮。种脊为一条纵沟,尖端有凹状的种脐。胚乳灰白色至黄白色。有特异香气,味辛、微苦。[/font] [b][font=宋体]【检查】 水分[/font][/b][font=宋体] [/font][font=宋体]同药材,不得过10.0%。[/font] [b][font=宋体]总灰分[/font][/b][font=宋体] [/font][font=宋体]同药材,不得过6.0%。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 同药材,含挥发油不得少于1.0%(ml/g)。[/font] [b][font=宋体]【鉴别】[/font][/b][font=宋体] 同药材。[/font] [b][font=宋体]姜草果仁[/font][/b][font=宋体] [/font][font=宋体]取净草果仁,照姜汁炙法(通则0213)炒干。用时捣碎。[/font] [font=宋体]本品形如草果仁,棕褐色,偶见焦斑。有特异香气,味辛辣、微苦。[/font] [b][font=宋体]【检查】 水分[/font][/b][font=宋体] [/font][font=宋体]同药材,不得过10.0%。[/font] [b][font=宋体]总灰分[/font][/b][font=宋体] [/font][font=宋体]同药材,不得过6.0%。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 同药材,含挥发油不得少于0.7%(ml/g)。[/font] [b][font=宋体]【鉴别】[/font][/b][font=宋体] 同药材。[/font] [b][font=宋体]【性味与归经】[/font][/b][font=宋体] 辛,温。归脾、胃经。[/font] [b][font=宋体]【功能与主治】[/font][/b][font=宋体] 燥湿温中,截疟除痰。用于寒湿内阻,脘腹胀痛,痞满呕吐,疟疾寒热,瘟疫发热。[/font] [b][font=宋体]【用法与用量】[/font][/b][font=宋体] 3[/font][font=宋体]~6g。[/font] [b][font=宋体]【贮藏】[/font][/b][font=宋体] 置阴凉干燥处。[/font]
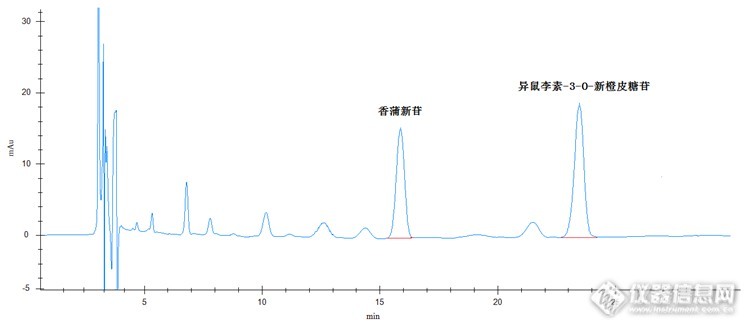
检测蒲黄药材快速、准确出结果 蒲黄药材具有止血,化瘀,通淋等药物功效。常用于治疗吐血,衄血,咯血,崩漏,外伤出血,经闭痛经,跌伤肿痛,血淋涩痛等疾病效果较好,属于名贵药材。 蒲黄里的药物成分很多,主要有异鼠李素-3-O-新橙皮糖苷、香蒲新苷。下面我们就针对这两种成分进行实验部分原理 取适量蒲黄药材,加纯甲醇溶解,超声波提取,经进样器进样,色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量计算。仪器及试剂仪器:高效液相色谱仪(紫外检测器+柱温箱),超声波清洗仪,溶剂过滤器,针筒式过滤器,电子天平试剂:甲醇(色谱纯),乙腈(色谱纯),磷酸溶液(分析纯),超纯水样品制备 对照品溶液的制备:准确称取异鼠李素-3-O-新橙皮糖苷、香蒲新苷对照品各2.5mg于50ml容量瓶中,加甲醇至刻度,配制成浓度均为50μg/ml的对照品溶液,备用。 供试品溶液的制备:准确称取本品约0.5g,置具塞锥形瓶中,精密加入50ml甲醇后,称定重量并记录,冷浸12小时后超声波超声30min,放冷,再次称定重量,用甲醇补足减少的重量,摇匀,滤过,待测。色谱条件检测器:紫外检测器色谱柱:普通C18,4.6 X 250mm,50μm流动相:乙腈:0.05%磷酸溶液=15:85(V:V)检测波长:254nm进样量20μl柱温:室温对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301828_558136_2536753_3.png供试品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301828_558137_2536753_3.png 从以上色谱图我们可以看出样品出峰时间较晚,峰形也较差。下面我们换用一根天津博纳艾杰尔科技有限公司生产的耐酸性色谱柱,效果我们请看色谱图。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2015/07/201507301828_558138_2536753_3.png 供试品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301829_558139_2536753_3.png 换了这根色谱柱,色谱图的峰形好了很多,保留时间也明显有所提前,但保留时间还是有点晚。下面我们把流动相比例调整了下,调整为乙腈:0.05%磷酸溶液=22:78(V:V),效果接着往下看。对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301829_558140_2536753_3.png供试品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301829_558141_2536753_3.png 当然保留时间还可以再缩短些(通过提高色谱柱温度,或提高高压泵流速,或换用更高效更短的色谱柱),但供试品中异鼠李素-3-O-新橙皮糖苷的附近有干扰物,为了保证分离度,这个分析应该时间比较合理(一定要根据样品情况而定,如果样品很纯净,对被测物没有干扰,保留时间当然是越短越好),尽量不要再缩短了,保证分离度是第一位的。 检测蒲黄药材的这个方法到现在已经比较完美。但有几点事项还需注意。1. 样
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font] (1)本品叶片近基部横切面:上表皮细胞类方形,壁厚,外被厚的角质层,主脉处有单细胞非腺毛;下表皮细胞略小,可见气孔。[color=var(--weui-LINK)]栅栏组织[i][/i][/color]为2~4列细胞,海绵组织疏松;主脉处上、下表皮内为1至数列[color=var(--weui-LINK)]厚角细胞[i][/i][/color]。主脉维管束外韧型,其上、下方均具木化纤维群。叶缘表皮内常依次为厚角细胞和[color=var(--weui-LINK)]石细胞[i][/i][/color]半环带,再内为木化纤维群;叶缘近叶柄处仅有数列厚角细胞,近基部以上渐无[color=var(--weui-LINK)]厚角组织[i][/i][/color]。叶缘表皮内和主脉处下表皮内厚角组织中偶有石细胞,韧皮部下方的纤维群外亦偶见。[color=var(--weui-LINK)]薄壁组织[i][/i][/color]和下表皮细胞常含草酸钙簇晶。 [font=宋体](2)取本品粉末2g,加70%乙醇40ml,超声处理30分钟,滤过,滤液蒸干,残渣加水40ml使溶解,加三氯甲烷40ml振摇提取,弃去三氯甲烷液,水层加浓氨试液2ml,摇匀,再加水饱和的正丁醇40ml振摇提取,分取正丁醇液,浓缩至干,残渣加甲醇2ml使溶解,作为供试品溶液。另取枸骨叶对照药材2g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各1[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体],分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(1:3:1:0.3)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体][/font] [font=宋体] [/font] [font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [b][font=宋体][/font] [font=宋体][/font] [font=宋体]水分[/font][/b][font=宋体] [/font][font=宋体]不得过8.0%(通则0832第二法)。[/font] [b][font=宋体]总灰分[/font][/b][font=宋体] [/font][font=宋体]不得过6.0%(通则2302)[/font]
下面是我曾经接手过的一个项目,内容是个人的经验,解释也不一定完全正确,大家参考了!!!小改进,大成功--药材TLC鉴别实例[b]关于××××丸成品鉴别方法的改进原有方法:[/b](1)取本品××g,加乙醚20ml,超声处理5分钟,滤过,药渣及滤纸一并置烧瓶中,加7%硫酸乙醇-水(1:3)混合液20ml,加热回流3小时,放冷,用氯仿振摇提取二次,每次20ml,合并氯仿液,加水30ml洗涤,弃去洗液,氯仿液用无水硫酸钠脱水,滤过,滤液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取桔梗对照药材1g,同法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取上述两种溶液各1~2µ l,分别点于同一硅胶G薄层板上,以氯仿-乙醚(1:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色斑点。(2) 取甘草次酸对照品,加甲醇制成每1 ml含1mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取[鉴别](1)项下的供试品溶液8µ l、对照品溶液5µ l,分别点于同一硅胶GF254薄层板上,以石油醚(30~60℃)-氯仿-冰醋酸(10:5:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[b]改进方法:[/b](1)取本品××g,加乙醚20ml,超声处理5分钟,滤过,药渣及滤纸再加氯仿30ml,超声10分钟,离心,药渣置烧瓶中,加氯仿30 ml和盐酸2ml,加热回流1小时,滤过,滤液蒸干,残渣加无水乙醇1ml使溶解,作为供试品溶液。另取桔梗对照药材0.25g,同法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取上述两种溶液各2~5µ l,分别点于同一硅胶G薄层板上,以氯仿-乙醚(1:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色斑点。(2) 取甘草次酸对照品,加甲醇制成每1 ml含1mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取[鉴别](1)项下的供试品溶液和对照品溶液各2~5µ l,分别点于同一硅胶GF254薄层板上,以石油醚(30~60℃)-氯仿-冰醋酸(10:5:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[b]关于××××丸鉴别方法的改进的说明[/b]原有方法对于样品和对照药材的提取存在着一定的问题,具体表现在如下几个方面:(1)对于甘草次酸的鉴别的机理说明:A乙醚的超声处理的目的:除去药材或浸膏中的酯溶性成分和制剂中大量存在的水溶性辅料,这些物质的存在直接影响薄层鉴别;B氯仿的超声处理的目的:除去药材、浸膏和制剂中的甘草次酸成分;C酸解的目的:通过酸解,可以使药材、浸膏和制剂中的甘草酸水解成为甘草次酸成分,这样可以与对照品进行比较;原有方法存在的不足:A加入酸的量不足,使得甘草酸水解不充分,只有少量甘草酸发生水解,致使该方法的重现性很差;B水解时间长,不适合企业的生产实际需要;C由于甘草次酸在氯仿和水中都有一定的溶解能力,无水硫酸钠脱水也可能包裹一定量的甘草次酸,所以原方法用氯仿萃取二次、加水30ml洗涤、无水硫酸钠脱水三项操作使得鉴别结果重现性很差,而且这些操作很费时,不同人员的操作的差异比较大。(2)对于桔梗的鉴别的机理说明:A乙醚的超声处理的目的:除去药材或浸膏中的酯溶性成分(如脂肪酸、脂肪油等)和制剂中大量存在的水溶性辅料,这些物质的存在直接影响薄层鉴别;B氯仿的超声处理的目的:除去药材、浸膏和制剂中的桔梗皂苷成分;C酸解的目的:通过酸解,可以使药材、浸膏和制剂中的桔梗皂苷(包括桔梗皂苷A、B、D2、D3,远志苷D、D2,桔梗皂酸A甲酯等)水解成为苷元(包括桔梗皂苷元、远志酸,桔梗酸等)成分,这样可以与对照药材进行比较;原有方法存在的不足:A加入酸的量不足,使得桔梗皂苷水解不充分,只有少量桔梗皂苷发生水解,致使该方法的重现性很差;B水解时间长,不适合企业的生产实际需要;C由于甘草次酸在氯仿和水中都有一定的溶解能力,无水硫酸钠脱水也可能包裹一定量的甘草次酸,所以原方法用氯仿萃取二次、加水30ml洗涤、无水硫酸钠脱水三项操作使得鉴别结果重现性很差,而且这些操作很费时,不同人员的操作的差异比较大。
西青果的薄层图?按药典方法提取展开,对照药材也没有斑点。是否有其他可行的方法?
天麻对照品天麻素和对羟基苯甲醇保留时间分别是11和22分钟,天麻药材出峰时间是15和26分钟,且时间还比较稳定,后面更换了新柱子,把柱温箱温度从30度调到35度,把泵从B泵更换到A泵,重新配流动相,供试品,发现还是一样漂移。而且之前做的特征图谱保留时间又对的上。流动相过滤了也超声了。真的不知道该咋办了??
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体](1)本品横切面:[b]温郁金[/b] 表皮细胞有时残存,外壁稍厚。根被狭窄,为4~8列细胞,壁薄,略呈波状,排列整齐。皮层宽约为根直径的1/2,油细胞难察见,内皮层明显。中柱韧皮部束与木质部束各40~55个,间隔排列;木质部束导管2~4个,并有微木化的纤维,导管多角形,壁薄,直径20~90μm。薄壁细胞中可见糊化淀粉粒。[/font] [b][font=宋体]黄丝郁金[/font][/b][font=宋体] [/font][font=宋体]根被最内层细胞壁增厚。中柱韧皮部束与木质部束各22~29个,间隔排列;有的木质部导管与纤维连接成环。油细胞众多。薄壁组织中随处散有色素细胞。[/font] [b][font=宋体]桂郁金[/font][/b][font=宋体] [/font][font=宋体]根被细胞偶有增厚,根被内方有1~2列[color=var(--weui-LINK)]厚壁细胞[i][/i][/color],成环,层纹明显。中柱韧皮部束与木质部束各42~48个,间隔排列;导管类圆形,直径可达160μm。[/font] [b][font=宋体]绿丝郁金[/font][/b][font=宋体] [/font][font=宋体]根被细胞无增厚。中柱外侧的皮层处常有色素细胞。韧皮部皱缩,木质部束64~72个,导管扁圆形。[/font] [font=宋体](2)取本品粉末2g,加无水乙醇25ml,超声处理30分钟,滤过,滤液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。另取郁金对照药材2g,同法制成对照药材溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(17:3)为展开剂,预饱和30分钟,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。置日光和紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的主斑点或荧光斑点。[/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体][/font][/b] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][b]水分[/b] 不得过15.0%(通则0832第四法)。[/font] [b][font=宋体]总灰分[/font][/b][font=宋体] [/font][font=宋体]不得过9.0%(通则2302)。[/font]
延胡索薄层样品比对照药材多了一个点,怎么回事?
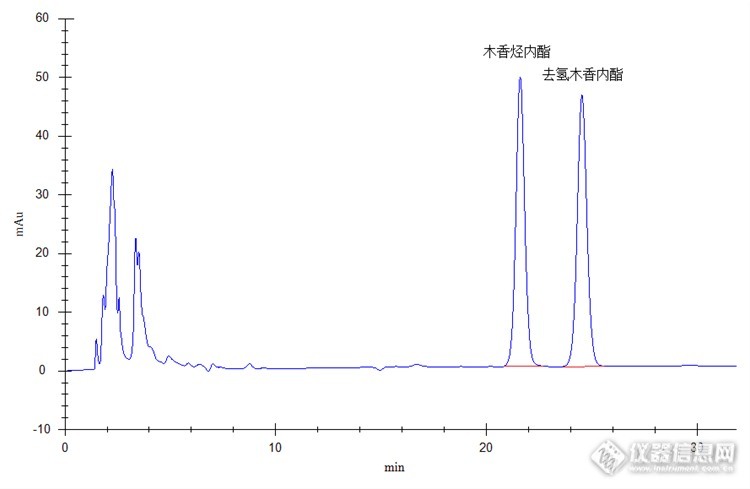
高效方法检测名贵药材 名贵药材很多种,有一种叫木香。 它花色艳丽,形状各异,香味迷人,漂亮极致。根、茎、叶也有着非常不菲的药用价值和营养成分。 药用部分主要来自根部,它气香特异,味微苦,含有丰富的去氢木香内酯,木香烯内脂,木香萜醛,木香内酯,二氢木香内酯,α-环木香烯内酯,β-环木香烯内酯,土木香内酯等药物成分。另外根部还含天冬氨酸,谷氨酸,甘氨酸,甘氨酸,瓜氨酸等20多种氨基酸,营养成分也非常丰富。 叶含蒲公英甾醇,α-香树精硬脂酸酯,β-香树精棕榈酸酯,羽扇醇棕榈酸酯等多种香精、香料成分。 木香具有行气、止痛、止血、健脾、消炎、抗菌、降压等多种药物功效,可用于多种疑难病症治疗。http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418455848_01_0_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418460455_01_0_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418462259_01_2536753_3.jpeghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508141901_560867_2536753_3.png说明:以上几张图片是网上截图,不是本人所拍,在这只是为了说明下木香药材的外观及美感,希望大家能够理解。 既然木香价值这么高,检测它就显得尤为重要,方法选择尤为关键。下面我们就介绍几种高效液相色谱法检测该药材中木香烃内酯、去氢木香内酯方法。1.实验部分1.1原理 精密称取该药品适量,超声波提取后经进样器注入高效液相色谱系统,通过C18色谱柱分离,紫外检测器检测,外标法(保留时间定性,峰面积定量)计算,得出所测成分含量。1.2 仪器及试剂 仪器:高效液相色谱仪(紫外检测器+高压恒流泵+柱温箱),四号筛(药典筛),超声波清洗器,溶剂过滤器,电子天平(0.0001级)等。 试剂:甲醇(色谱纯),超纯水1.3样品制备 对照品溶液制备:准确称取木香烃内酯对照品、去氢木香内酯对照品各5mg于50ml容量瓶中,加甲醇至刻度,配制成含木香烃内酯对照品、去氢木香内酯各0.1mg/ml的混合对照品溶液,备用。 供试品溶液制备:取本品适量充分粉碎后过四号筛,准确称取过筛粉末0.3g,置于具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,放置过夜后超声处理30分钟,放冷,再次称定重量,用甲醇补足减少的重量,摇匀,0.45μm微膜滤过,待测。1.4 色谱条件检测器:紫外检测器检测波长:225nm色谱柱:Promosil C18,4.6 X 250mm,5μm流动相:甲醇-水(65:35)流速:1.0ml/min柱温:室温进样量:10μl2、实验过程及色谱图对照品色谱图http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418542201_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418545304_01_2536753_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418542563_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418545925_01_2536753_3.png 计算公式:样品中各物质的含量计算公式:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418541866_01_2536753_3.pngD----供试品中被测物含量,以(%)表示V----供试品稀释的总体积,单位(mL)P3----供试品溶液被测物峰面积值P4-----对照品溶液被测物峰面积值ρ2----对照品被测物浓度。单位为mg/mlm2----样品质量,单位(mg) 通过计算木香烃内酯的含量大概是0.8%,去氢木香内酯的含量大概是1.6%,总含量2.4%,远大于药典中要求的1.8%,该木香药材满足药典要求,属合格品。 这个检测时间有点长,我们不防改变下色谱条件,缩短些时间。那就把柱温调整到40℃,看下结果。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418542990_01_2536753_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418543464_01_2536753_3.png 看来有效果,检测时间是短了,但分离度也相对差了点,不够理想。那我们再不防改变下色谱条件,争取把分离度效果弄好些。那就调整下流动相,甲醇-水(60:40),看下结果。对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418544178_01_2536753_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/201508141854474
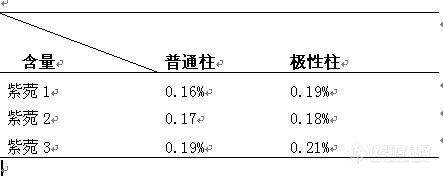
中药分析版面原创不太活跃,我小搞一下,希望大家踊跃参与~!七月底最后赶上!这段时间太忙了,呵呵!我在这里抛砖引玉,由于时间匆忙,可能会有纰漏,望大家多提宝贵意见!哈哈。。。。运用不同色谱柱测定紫菀含量的研究 目的:比较不同类型色谱柱对紫菀含量测定的影响。方法:按照10版药典方法,我们分别运用了常规色谱柱和极性色谱柱测定紫菀中紫菀酮的含量。结果:二者测定紫菀酮均可,但极性色谱柱稍优于普通柱。结论:极性柱可以作为紫菀测定中用到的色谱柱。 紫菀为菊科植物紫菀Aster tataricus L.f.的干燥根和根茎。春、秋二季采挖,除去有节的根茎(习称“母根”)和泥沙,编成辫状晒干,或直接晒干。国内主产于河北、内蒙和东北三省,通常生长于潮湿的河边地带,是一味著名中药,有止咳祛寒之功效。根含紫菀酮(shionone)、槲皮素、无羁萜、表无羁萜和挥发油,尚含紫菀皂甙(astersaponin),水解得常春藤皂甙元(hederagenin)。 一 仪器与试药 (一)仪器 Dionex 3000 高效液相色谱系统 DIONEX 普通色谱柱 Dionex 极性色谱柱 KQ250超声波清洗器(江苏昆山超声仪器厂) 高速离心机(上海飞鸽) 分析天平(梅特勒公司)。 (二)试药 紫菀药材:所有药材均由提供。 对照品:紫菀酮(对照品) 化学试剂:乙腈为色谱纯;甲醇(分析纯,色谱纯);水为重蒸水。 二、实验方法与结果 (一)按照2010版药典紫菀药材测定下制备供试品 (二)运用不同色谱柱,按2010版药典测定紫菀药材中紫菀酮的含量 (三)测定结果见下表和下图。http://ng1.17img.cn/bbsfiles/images/2011/08/201108011048_307675_2961690_3.jpg 1.普通柱紫菀酮HPLC图http://ng1.17img.cn/bbsfiles/images/2011/08/201108011048_307676_2961690_3.jpg2.普通柱供试品色谱峰图http://ng1.17img.cn/bbsfiles/images/2011/08/201108011049_307677_2961690_3.jpg3.极性柱紫菀酮色谱峰图http://ng1.17img.cn/bbsfiles/images/2011/08/201108011049_307678_2961690_3.jpg4.极性柱供试品色谱峰图含量测定见下表http://ng1.17img.cn/bbsfiles/images/2011/08/201108011049_307679_2961690_3.jpg三、小结与讨论(一)2010版药典测定紫菀中紫菀酮含量的方法是以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(96:4)为流动相;检测波长为200nm;柱温40℃。理论板数按紫菀酮峰计算应不低于3500。但由于所选波长在末端吸收,HPLC图谱效果不是很好。(二)根据紫菀酮的性质特点,为了进一步改善紫菀酮测定条件,我们选用了极性色谱柱来代替常规色谱柱,HPLC图谱较为理想,和常规色谱柱测定结果相近,误差较小。(三)极性色谱柱可用作紫菀含量测定,为进一步完善紫菀药材含测研究奠定了基础。
[size=3]根据口尝中药材来体会其特殊味道和口感,从而衡量和鉴别药材真伪优劣的方法,称口试法。该法简便易行,对鉴别根皮类、果实种子类等中药材有较大适用价值。 一、有些药材具有鲜明纯正而恒久的苦、酸、甜、咸、辣等味,且一般味道越浓,质量越好。 黄连、苦参、山豆根、穿心莲、胡黄连、苦杏仁、鸦胆子味道极苦,且越苦越佳;乌梅、木瓜、山楂以味酸为好;蜂蜜、甘草、党参、罗汉果以味甜为好;全蝎、土元味咸;干姜、郁金、高良姜、草果、草豆蔻味辛辣等。 二、有些中药材药味间杂,同时具有两种以上味道,或嚼之稍久而变味。 黄芪、沙苑子嚼之味甜而有豆腥气;西洋参、陈皮、板蓝根、桔梗先甜而后苦;枳壳先苦而后微酸;续断味苦、微甜而后涩;厚朴苦而辛辣;肉桂嚼之味甜辣,渣少为佳;秦艽、双边栝楼根味苦涩等。 三、有些药材根据口味和口感可资鉴别。 知母、石斛、麦冬、鹤虱、白及微甜又略苦,嚼之有黏性;黄精、玉竹、白术味甜有黏性;山药味微酸,嚼之发黏;天南星、三棱、蛇床子、牵牛子、牡丹皮、徐长卿口尝有麻辣味乃至麻舌感;苏合香、鹿角霜、茯苓、龙骨、天竺黄嚼之发黏;雷丸嚼之初有颗粒感,微带黏性,久嚼无渣;冰片、白豆蔻味辛凉浓烈;蛤蟆油嚼之有黏滑感;大黄嚼之发黏,有沙粒感,唾液染成黄色;黄柏味苦,嚼之有黏液性,唾液染成黄色;枸杞子嚼之味甜,唾液呈红黄色;杜仲味微苦,嚼之有胶状感;乌药味微苦,有清凉感;木香味苦辛,但川木香却又嚼之发黏;胖大海、车前子嚼之均有黏液性等。 四、有些药材口尝时有明显或强烈的刺激性。 合欢皮味微涩、稍刺舌,而后喉头有不适感;白附子、半夏嚼之麻辣而刺喉、刺舌;藜芦粉味苦,有强烈的催嚏性,对舌有较强刺激性和烧灼感;远志有一种特殊的苦味,伴刺喉感等。[/size]
根据口尝中药材来体会其特殊味道和口感,从而衡量和鉴别药材真伪优劣的方法,称口试法。该法简便易行,对鉴别根皮类、果实种子类等中药材有较大适用价值。 一、有些药材具有鲜明纯正而恒久的苦、酸、甜、咸、辣等味,且一般味道越浓,质量越好。 黄连、苦参、山豆根、穿心莲、胡黄连、苦杏仁、鸦胆子味道极苦,且越苦越佳;乌梅、木瓜、山楂以味酸为好;蜂蜜、甘草、党参、罗汉果以味甜为好;全蝎、土元味咸;干姜、郁金、高良姜、草果、草豆蔻味辛辣等。 二、有些中药材药味间杂,同时具有两种以上味道,或嚼之稍久而变味。 黄芪、沙苑子嚼之味甜而有豆腥气;西洋参、陈皮、板蓝根、桔梗先甜而后苦;枳壳先苦而后微酸;续断味苦、微甜而后涩;厚朴苦而辛辣;肉桂嚼之味甜辣,渣少为佳;秦艽、双边栝楼根味苦涩等。 三、有些药材根据口味和口感可资鉴别。 知母、石斛、麦冬、鹤虱、白及微甜又略苦,嚼之有黏性;黄精、玉竹、白术味甜有黏性;山药味微酸,嚼之发黏;天南星、三棱、蛇床子、牵牛子、牡丹皮、徐长卿口尝有麻辣味乃至麻舌感;苏合香、鹿角霜、茯苓、龙骨、天竺黄嚼之发黏;雷丸嚼之初有颗粒感,微带黏性,久嚼无渣;冰片、白豆蔻味辛凉浓烈;蛤蟆油嚼之有黏滑感;大黄嚼之发黏,有沙粒感,唾液染成黄色;黄柏味苦,嚼之有黏液性,唾液染成黄色;枸杞子嚼之味甜,唾液呈红黄色;杜仲味微苦,嚼之有胶状感;乌药味微苦,有清凉感;木香味苦辛,但川木香却又嚼之发黏;胖大海、车前子嚼之均有黏液性等。 四、有些药材口尝时有明显或强烈的刺激性。 合欢皮味微涩、稍刺舌,而后喉头有不适感;白附子、半夏嚼之麻辣而刺喉、刺舌;藜芦粉味苦,有强烈的催嚏性,对舌有较强刺激性和烧灼感;远志有一种特殊的苦味,伴刺喉感等。 在使用口试鉴别法时要注意两点:一是取样要有代表性,药材各部位的味觉可能不同;二是对强烈刺激性和剧毒药材,口尝时要小心,取样要少,尝后要立即吐出,漱口,洗手,以防中毒。另外有些中药材如斑蝥因刺激性强,不宜口尝。特别说明:非专业人士请勿轻易随便口试中药材,以免中毒!!!
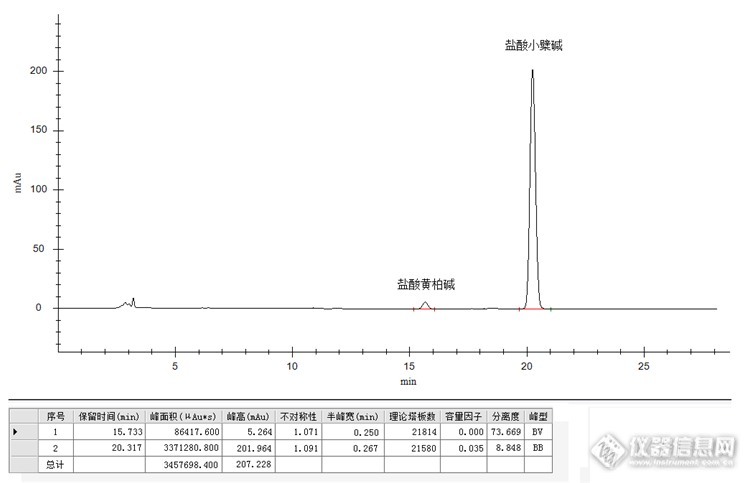
药材黄柏中多种成分检测 黄柏是一种中药材,属于芸香科植物黄皮树的干燥树皮。味极苦,略咸。具有清热燥湿、泻火除蒸、解毒疗疮、滋阴降火等药物疗效。是治疗湿热泻痢、带下阴痒、涩痛火旺、脚气痿躄、劳热躁汗、阴虚遗精、疮疡肿毒、湿疹湿疮等病症良药。 黄柏中含有丰富的药物成分,其中以盐酸小檗碱和盐酸黄柏碱为主。这两种成分也是药典中明确要求检测的项目。同一药材中的这两种成分药典检测方法还不一样,下面我们就来看看。 高效液相色谱法检测药材黄柏中盐酸小檗碱和盐酸黄柏碱含量。实验部分【原理】 精密称取适量黄柏样品经流动相溶解,超声提取后注入高效液相色谱系统,C18色谱柱分离,紫外检测器检测,外标法(保留时间定性,峰面积定量)计算,得出所测成分含量。【仪器及试剂】 仪器:高效液相色谱仪(紫外检测器+等度泵+柱温箱+自动进样器+在线脱气机等),超声波清洗仪,溶剂过滤器,电子天平(0.0001),药典筛(三号筛、四号筛)等。 试剂:乙腈(色谱纯),磷酸(分析纯),十二烷基磺酸钠(优级纯),超纯水等。 这两种成分的结构及特性及药材中的含量等差异可能有些大,药典中的方法是分开检测的,下面我们先来检测黄柏中盐酸小檗碱。【样品制备】 对照品溶液制备:精密称取盐酸小檗碱对照品2.5mg于25ml容量瓶中,加流动相溶解、定容,制成0.1mg/ml盐酸小檗碱对照品溶液,备用。 供试品溶液制备:取黄柏药材适量,充分粉碎后过药典筛三号筛,精密称取过筛后粉末0.1g置于100ml容量瓶中,加流动相80ml,超声处理40分钟,放冷,用流动相稀释至刻度,摇匀,0.45um有机相微膜滤过,待测。【色谱条件】检测器:紫外检测器检测波长:265nm色谱柱:Promosil C18 ( 250 mm X 4.6mm,5μm )流动相:乙腈:0.1%磷酸溶液(每100ml磷酸溶液中加十二烷基磺酸钠0.2g)=50:50(V:V)流速:1.0ml/min柱温:30℃进样量:5μl【色谱图】对照品溶液色谱图: http://ng1.17img.cn/bbsfiles/images/2015/08/201508312252_563822_2536753_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/08/201508312252_563823_2536753_3.png 下面我们再来检测黄柏中盐酸黄柏碱。 原理、仪器及试剂和上面一样,在此略过。【样品制备】 对照品溶液制备:精密称取盐酸黄柏碱对照品2.5mg于25ml容量瓶中,加流动相溶解、定容,制成0.1mg/ml盐酸黄柏碱对照品溶液,备用。 供试品溶液制备:取黄柏药材适量,充分粉碎后过药典筛四号筛,精密称取过筛后粉末0.5g置于具塞锥形瓶中,加流动相25ml,称定重量,超声处理30分钟,放冷,再次称定重量,用流动相补足减少的重量,摇匀,0.45um有机相微膜滤过,待测。【色谱条件】检测器:紫外检测器检测波长:284nm色谱柱:Promosil C18 ( 250 mm X 4.6mm,5μm )流动相:乙腈:0.1%磷酸溶液(每100ml磷酸溶液中加十二烷基磺酸钠0.2g)=36:64(V:V)流速:1.0ml/min柱温:30℃进样量:5μl【色谱图】对照品溶液色谱图: http://ng1.17img.cn/bbsfiles/images/2015/08/201508312253_563824_2536753_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/08/201508312253_563825_2536753_3.png 药典这个方法有些麻烦,对于成分不是特别复杂的药材大可不必这样,两个方法合并成一个方法完全可以搞定。下面我们就来看看。 原理、仪器及试剂和上面一样,在此略过。【样品制备】 对照品溶液制备:精密称取盐酸小檗碱和盐酸黄柏碱对照品各2.5mg于25ml容量瓶中,加流动相溶解、定容,制成0.1mg/ml盐酸小檗碱和盐酸黄柏碱混合对照品溶液,备用。 供试品溶液制备:取黄柏药材适量,充分粉碎后过药典筛三号筛,精密称取过筛后粉末0.5g置于100ml容量瓶中,加流动相至刻度,称定重量,超声处理30分钟,放冷,再次称定重量,用流动相补足减少的重量,摇匀,0.45um有机相微膜滤过,待测。【色谱条件】检测器:紫外检测器检测波长:序号时间(min)波长(nm)10285217285317.1265430265色谱柱:Promosil C18 ( 250 mm X 4.6mm,5μm )流动相:乙腈:0.1%磷酸溶液(每100ml磷酸溶液中加十二烷基磺酸钠0.2g)=40:60(V:V)流速:1.0ml/min柱温:30℃进样量:5μl【色谱图】对照品溶液色谱图: http://ng1.17img.cn/bbsfiles/images/2015/08/201508312253_563826_2536753_3.png供试品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2015/08/201508312253_563827_2536753_3.png 这样一个方法很方便的就把两种成分检测了出来,效果还不错。但由于这两种成分在该条件下的响应值和在样品中的含量有很大的差异,这样就导致一个色谱峰很大另一个偏小,对检测结果和色谱效果都有一定影响。 这样检测时我们就得考虑样品中多种成分在同一条件下的响应值及各自的含量,这两个因素如果差异不大,用同一种检测方法检测完全没问题。但如果差异较大,实验对结果准确度要求又不太高,这种方法就非常有优势,检测方便、快速。但如果对结果的准确度要求很高,这种方法最好别用,以防得不偿失。 同一样品检测方法很多,各有优势,实验时我们一定得立足现状,充分考虑样品及实验具体情况,满足实验要求是第一位的,切不可盲目的追求快而丢了准,或是盲目的追求准而浪费了时间、经费和精力等。 实验要求第一位,切记! 以上是个人点小小的心得,仅供参考。
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font][/b][font=宋体][/font][b][font=宋体][/font][/b] [font=宋体] [/font] [font=宋体][/font] [font=宋体](1)本品根横切面:木栓细胞6~12列,含棕色物。栓内层薄壁细胞有的含红棕色颗粒。韧皮部细胞较小。形成层不甚明显。木质部占根的主要部分,全部木化,射线不明显。薄壁细胞含草酸钙针晶束。[/font] [font=宋体](2)取本品粉末0.2g,加乙醚5ml,振摇数分钟,滤过,滤液加氢氧化钠试液1ml,振摇,静置使分层,水层显红色;醚层无色,置紫外光灯(365nm)下观察,显天蓝色荧光。[/font] [font=宋体](3)取本品粉末0.5g,加甲醇10ml,超声处理30分钟,滤过,滤液浓缩至1ml,作为供试品溶液。另取茜草对照药材0.5g,同法制成对照药材溶液。再取大叶茜草素对照品,加甲醇制成每1ml含2.5mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各5[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体],分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-丙酮(4:1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点。[/font] [font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size][b][font=宋体][/font][/b] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体]水 分 [/font][/b][font=宋体] [/font][font=宋体]不得过12.0%(通则0832第二法)。[/font] [b][font=宋体]总灰分 [/font][/b][font=宋体] [/font][font=宋体]不得过15.0%(通则2302)。[/font] [b][font=宋体]酸不溶性灰分[/font][/b][font=宋体] [/font][font=宋体]不得过5.0%(通则2302)。[/font] [b][font=宋体]【浸出物】[/font][/b][font=宋体] 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用乙醇作溶剂,不得少于9.0%。[/font] [b][font=宋体]【含量测定】 [/font][/b][font=宋体]照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以十八烷基硅烷键合硅胶为填充剂;以甲醇-乙腈-0.2%磷酸溶液(25:50:25)为流动相;检测波长为250nm。理论板数按大叶茜草素、羟基茜草素峰计算均应不低于4000。[/font] [b][font=宋体]对照品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取大叶茜草素对照品、羟基茜草素对照品适量,精密称定,加甲醇分别制成每1ml含大叶茜草素0.1mg、含羟基茜草素40[/font][font=&]μ[/font][font=宋体]g[/font][font=宋体]的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备 [/font][/b][font=宋体]取本品粉末(过二号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇100ml,密塞,称定重量,放置过夜,超声处理(功率250W,频率40kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液50ml,蒸干,残渣加甲醇-25%盐酸(4:1)混合溶液20ml溶解,置水浴中加热水解30分钟,立即冷却,加入三乙胺3ml,混匀,转移至25ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]分别精密吸取对照品溶液10[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体]与供试品溶液20[/font][font=&]μ[/font][font=宋体]l[/font][font=宋体],注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,即得。[/font] [font=宋体]本品按干燥品计算,含大叶茜草素([/font][font=&]C[sub]17[/sub]H[sub]15[/sub]O[sub]4[/sub][/font][font=宋体])不得少于0.40%,羟基茜草素([/font][font=&]C[sub]14[/sub]H[sub]8[/sub]O[sub]5[/sub][/font][font=宋体])不得少于0.10%。[/font] [font=宋体] [/font]
如题 中药材野生的跟种植的疗效有区别吗
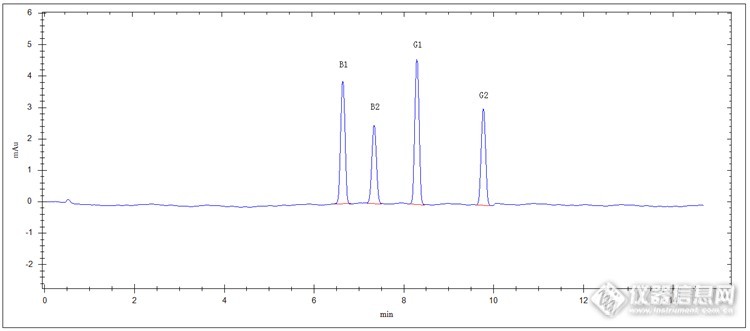
中药材中黄曲霉毒素检测方案 黄曲霉毒素是一类真菌(如黄曲霉和寄生曲霉)的有毒代谢产物,它们具有很强的致癌性,主要存在于药材、谷物、坚果、花生、瓜子、棉籽以及动物饲料等相关的产品中。其毒性远远高于氰化物、砷化物和有机农药毒性,其中以B1毒性最大。人摄入含有该类毒素物品时,摄入量在身体内是要累积的,不管量大量小对身体都有较大伤害。 近年来,各级食品药品等监管部门不断加大生产流通监管力度,努力保持食药多环节总体质量。现在对乳制品、玉米等食品及食品原料都有相关检测标准,2015版药典也重点明确该项目的检测。 下面我们就一起分享一下高效液相色谱法柱后光化学衍生荧光检测器检测中药材中黄曲霉毒素方法等重要内容。实验原理 样品经甲醇-水溶解,搅拌、离心及免疫亲和柱层析净化提取,进入高效液相色谱系统,亲水性C18色谱柱分离,经光化学衍生器柱后衍生,紫外检测器检测,外标法计算,即得。仪器与试剂 仪器:高效液相色谱仪(荧光检测器+等度泵+亲水性C18色谱柱+光化学衍生器等),气控操作架(含气泵),固相萃取装置,黄曲霉毒素免疫亲和柱,高速搅拌仪(搅拌速度大于11000转/分钟),离心机(离心速度4000转/分钟),氮吹仪,超声波清洗器,溶剂过滤器,电子天平等。 试剂:黄曲霉毒素对照品(均为SIGMA标准品),甲醇(色谱纯),氯化钠(分析纯),超纯水等。样品制备 混合对照品溶液制备:精密量取黄曲霉毒素混合标准液(黄曲霉毒素B1、黄曲霉毒素B2、黄曲霉毒素G1、黄曲霉毒素G2,标示浓度分别为1.0μg/ml、 0.3μg/ml、1.0μg/ml、0.3μg/ml)0.5ml,置于10ml容量瓶中,用甲醇稀释至刻度,制备为标准品储备液。精密量取储备液1ml,置于25ml容量瓶中,用甲醇稀释至刻度,备用。 供试品溶液制备:取适量被测某药材(或药品)充分粉碎,过二号筛(药典筛),精密称取过筛粉朱15g,加入氯化钠3g,置于具塞锥形瓶中,精密加入70%甲醇溶液75ml,高速搅拌仪搅拌2分钟,离心机离心5分钟,精密量取上清液15ml,置50ml容量瓶中,用水稀释至刻度,摇匀,微膜(0.45μm)滤过,精密量取续滤液20.0ml,通过固相萃取装置萃取(免疫亲合柱),流速每分钟3ml,用水20ml洗脱,洗脱液弃去,使空气进入柱子,将水挤出柱子,再用5ml甲醇洗脱,收集洗脱液,氮吹仪吹至近干,用甲醇定容1ml,摇匀,微膜(0.45μm)滤过,待测。色谱







 我要推广仪器
我要推广仪器
 下载APP
下载APP