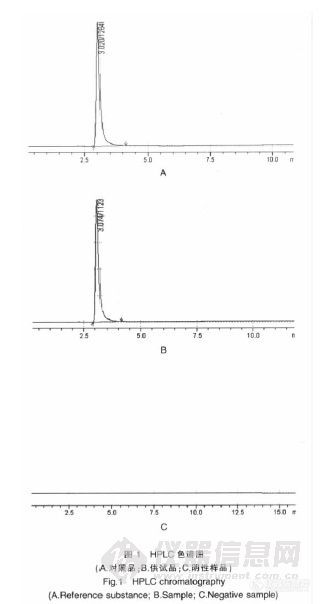
作者:康军;李征; (辽阳市食品药品检验所;)摘要:目的:建立高效液相法测定盐酸吗啉胍片中盐酸吗啉胍含量的方法。方法:采用Diamonsil C18(4.6 mm×200 mm,5μm)色谱柱;流动相:甲醇-乙腈-磷酸盐缓冲液(60∶20∶20);检测波长:237 nm;流速:1.0 ml/min。结果:盐酸吗啉胍在20~70μg范围内与峰面积呈良好的线性关系(r=0.999 2),平均回收率为99.90%,RSD=0.69%(n=9)。结论:本方法简便、准确、重现性好,可用于盐酸吗啉胍片中盐酸吗啉胍的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208201408_384677_1606903_3.jpg
谁有盐酸吗啉胍和乙酸铜的检测方法
请教各位大侠,检测盐酸吗啉胍大家都用的什么柱子,国标要求的色谱条件是氨基键合硅胶柱,流动相:0.03mol/L磷酸二氢钾缓冲液(PH4.5)。请做过的大侠帮忙解答下。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102920]动物组织中盐酸吗啉胍残留的高效液相色谱-串联质谱测定[/url]
哪位大侠,有优级纯(GR,Guaranteed reagent)、分析纯(AR,Analytical reagent、化学纯(CP,Chemical pure)盐酸的杂质含量标准,小弟十分感谢![em09511]
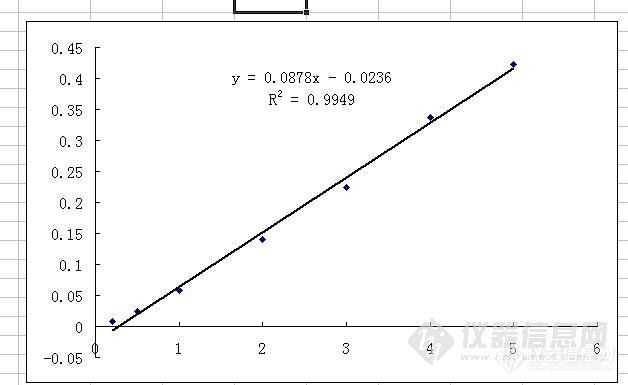
本人做了4天氰化物标准曲线了,采用的是HJ 484-2009异烟酸吡唑啉酮分光光度法,4次做出来的标准曲线都是中间几个点吸光度值明显偏低,重新配制过试剂,也用过30℃水浴锅恒温,还做过人员比对,做出来结果基本都是中间几个点吸光度值明显偏低,今天的测试结果为:空白0.015,扣除空白后0.2ug点吸光度0.008,0.5ug点吸光度0.024,1.0ug点吸光度0.058,2.0ug点吸光度0.140,3.0ug点吸光度0.224,4.0ug点吸光度0.337,5.0ug点吸光度0.423,今天测的空白有点高,但是前3天空白都是0.004和0.005。有没有做过异烟酸吡唑啉酮法测氰化物标准曲线的大虾,帮忙分析下原因,急啊!先谢谢了!http://ng1.17img.cn/bbsfiles/images/2010/11/201011201038_260810_1644065_3.jpg俺帮您做了一个曲线,这样大家看着方便点(二虎)
1. 可用作医药、农药、染料及其他有机合成中间体。可用来合成2-氨基嘧啶、2-氨基-6-甲基嘧啶、2-氨基-4,6-二甲基嘧啶,是制造磺胺嘧啶、磺胺甲基嘧啶、磺胺二甲基嘧啶等磺胺药物的中间体。 2. 盐酸胍(或硝酸胍)与氰乙酸乙酯反应,环合为2,4-二氨基-6-羟基嘧啶,用于合成抗贫血药叶酸。还可用作合成纤维的防静电剂。 3. 也可用于蛋白质变性剂。化学性质如下:1.性状:白色或微黄色块状物2.熔点(℃):181-1833.相对密度(g/mL,20/4℃):1.3544.溶解性:在20℃时在100g水中可以溶解228g,在100g甲醇中可以溶解76g,在100g乙醇中可以溶解24g。几乎不溶于丙酮、苯和乙醚。5.pH 值(4%水溶液,25℃):6.4
食品安全国家标准 食品营养强化剂 L-盐酸赖氨酸
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测环丙沙星,但标准品买的是盐酸环丙沙星,在配置时需要扣掉盐酸的质量吗?另外我用乙腈溶解发现溶解后的溶液白色浑浊,很多白色的悬浮物,加100ul甲酸后仍然浑浊,老师们有遇到过这种情况吗?
求教盐酸胍的分析方法,当然是越详细越好。谢谢
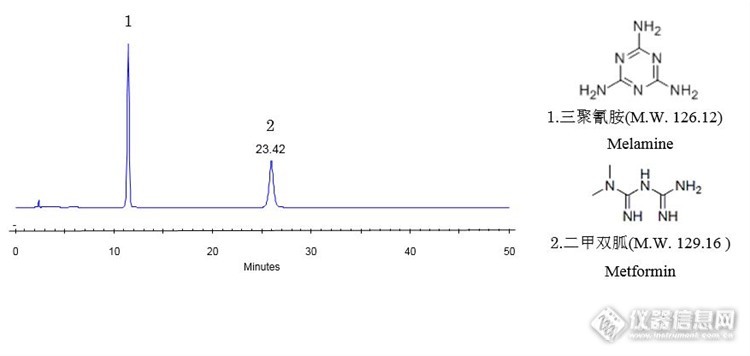
在9月29日的帖子中,我们介绍了在强阳离子交换模式下,使用CAPCELL PAK SCX UG80色谱柱对双胍类化合物进行保留与分离,并且尝试缩短分析时间的实验分析例。(详见 http://bbs.instrument.com.cn/topic/6289670)今天将要为大家带来的是使用该色谱柱,按照2015年版《中国药典》方法对盐酸二甲双胍进行的分析数据。盐酸二甲双胍(Metformin HCL)为白色结晶性粉末,无臭。在水中易溶,在甲醇中溶解,在乙醇中微溶,在氯仿或乙醚中不溶。熔点为220~225℃。分子式:C4H12ClN5分子量:165.6246以下为使用资生堂强阳离子交换色谱柱CAPCELL PAK SCX UG80对盐酸二甲双胍检测得到的谱图,请参考。http://ng1.17img.cn/bbsfiles/images/2016/10/201610271024_615216_2222981_3.jpg色谱柱:CAPCELL PAK SCX UG80 ;4.6mm i.d.×250mm流动相:1.7%W/V磷酸二氢铵(磷酸调pH至 3.0)流 速:1.0mL/min温 度:40 °C检 测:PDA 218nm进样量:20 µL样 品:5 µg/mL(按照药典方法配置)*注:峰上标数字为分离度。
总碱度测定时用盐酸滴定试样,看的是pH值从8.3滴定至4.5消耗的盐酸标准溶液的体积吗?还是要加上酚酞碱度消耗的盐酸标准溶液的体积?
各位老师,关于71-3中0.07mol/L 的盐酸标定,有国标标准时说关于盐酸标定的么?
原料药盐酸米多君标准(国内:YBH09452006),5-氨基乙酰丙酸盐酸盐标准。若有特殊要求也可站内信息联系!有国外标准也行。
实验室盐酸标准溶液通常都是用玻璃瓶来装的,如果配好的标准溶液需要搬到其它实验室用,为了搬运过程安全,可以用塑料壶来装吗?
这个问题涉及到很多方面,故放在综合区,望版主谅解。近日在考察注射用盐酸阿糖胞苷是否可用USP37的标准。发现一个问题,USP37收载的该剂型的活性物质是阿糖胞苷,而不是盐酸阿糖胞苷;而在查阅国外的几个制药公司的该产品的说明书,注明的活性物质也是阿糖胞苷原型,而非盐酸盐。故在此想请教对药物化学和药物分析很了解的童鞋们几个问题:1、国外的该产品说明书上注明的是阿糖胞苷原型药,USP37收载的也是原型药,是否说明在国外该制剂采用的原料药是阿糖胞苷,而非其盐酸盐?2、国内的制药厂家的该产品说明书上注明的活性物质是盐酸阿糖胞苷,且中国药典收载的也是盐酸阿糖胞苷,毫无疑问说明原料药采用的是盐酸阿糖胞苷,为什么要用盐酸盐,而不跟国外一样采用原型药?是为了避开专利还是盐酸盐的形式更有利于人体吸收?3、USP37收载原型药,被检测的药物是盐酸盐,是否说明该药物不能用USP37的标准来检?4、在哪里可以查到国外药物的专利,及其详细信息,如药物的化学结构、化学式、其专利到期的时间等?5、如何可以准确地知道国外一些药品制剂所使用的原料的详细信息?尤其是理化方面的信息,这些信息在说明书上体现的很少。望高手不吝赐教!谢谢!
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']盐酸中甲苯测定,可以直接萃取后进样分析吗?标准样品配制需要在盐酸中加入不同量的甲苯配制吗?[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
GB/T6432-94标准中4.6 盐酸标准溶液:邻苯二甲酸氢钾法标定,按GB601制备。平常都是用碳酸钠标定盐酸,用邻苯二甲酸氢钾标定氢氧化钠,这里却.......该如何理解呢?
本人在此急求 中华人民共和国国家药品监督管理局标准(试行)中的关于"盐酸左氧氟沙星注射液"的标准,请大家帮忙!谢谢!
我听说现在盐酸不能作为标准溶液了,不知道对不对,请指教下。谢谢
急!!!!谁有氰化氢的标准曲线图(异烟酸-吡唑啉酮分光光度法)
我目前急需副产品盐酸标准 HG/T3783,网站搜索不到。请各位帮忙提供下。感激不尽!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
请教各位一下:在标定盐酸标准溶液时,用无水碳酸钠作基准准确还是用硼砂准确? 假如我要配制0.5mol/L的盐酸,标出的结果是0.4741mol/L,溶液的体积估计为13000mL,请问我应该加几毫升浓盐酸? 谢谢
盐酸二甲双胍格列本脲胶囊I有关物质按照15版药典配制双氰胺,三聚氰胺,和二甲双胍的出峰时间顺序是什么,求分享
大神们,有做过食品中烟酸和烟酸胺的测定的么,GB5009.89-2016.要参加中检院的能力比对奶粉中烟酸和烟酸胺的测定,第一次接触这个项目,有什么需要注意的地方,包括溶剂哪个厂家好,柱子哪家的出峰效果好,标准溶液买哪里的等等。。。。请做过此项目的同学留下你们宝贵滴经验,先谢谢啦!!

盐酸二甲双胍格列本脲胶囊I有关物质按照2015版药典配制,三聚氰胺不出峰,色谱柱用的资生堂的。求各位大神指点一下[img]https://ng1.17img.cn/bbsfiles/images/2019/05/201905021326407220_6369_3384811_3.jpg[/img]
请问环保部的氯化物标准液,能否作为测定盐酸的标准物质呢!!!
1mol/L盐酸标准溶液在使用半个月后浓度变高是怎么回事呢?我是用无水碳酸钠标定的,如果说盐酸挥发了,那浓度应该是变低,可现在变高了是怎么回事呢?大概高了0.1左右,请教各位大侠了,谢谢!

[img=,690,75]https://ng1.17img.cn/bbsfiles/images/2020/01/202001161621460228_8391_3269810_3.png!w690x75.jpg[/img]这个标准所写的异烟酸-吡唑啉酮配制方法是完全没有异烟酸的加入,那这个试剂就不是完整的了,不能当做显色剂,正确的方法该如何配制?水质HJ484的标准里面有写,异烟酸-吡唑啉酮是先将1.5g异烟酸溶于25ml 20g/L的100ml,然后0.25g吡唑啉酮溶于20ml的甲酰胺里,再按吡唑啉酮和异烟酸1:5混合。首先按称取量来说的话,称取量不一样,海水的吡唑啉酮量比较大,如果我单纯用水质的显色剂配制用作海水的显色会不会令显色不完全导致精度变低;那如果我按照1.0g吡唑啉酮溶于40ml甲酰胺中,相对的我异烟酸的比例是否需要等比例增加?但海水标准只说了两液合并于100ml容量瓶里面,需要加水至标线,没有一个两者的比例要求,我如何确定比例?12763.4里面没有关于海水氰化物的相关标准。







 我要推广仪器
我要推广仪器
 下载APP
下载APP